•
Author: Christophe Dessimoz •
∞
Our latest paper, “Standardized Benchmarking in the Quest for Orthologs”, just came out in Nature Methods. This is a brief overview and story behind the paper.
Orthology benchmarking
Orthology, which formalises the concept of “same” genes in different species, is a foundation of genomics. Last year alone, more than 13,000 scientific papers were published with keyword “ortholog”. To satisfy this enormous demand, many methods and resources for ortholog inference have been proposed. Yet because orthology is defined from the evolutionary history of genes, which usually cannot be known with certainty, benchmarking orthology is hard. There are also practical challenges in comparing complex computational pipelines, many of which are not available as standalone software.
Identifier mapping: the bane of bioinformatics
Back in 2009, Adrian Altenhoff and I published a paper on ortholog benchmarking in PLOS Computational Biology. At the time, this was the first benchmark study with phylogeny-based tests. It also investigated an unprecedented number of methods. One of the most challenging aspect of this work—and by far the most tedious—was to compare inferences performed by different methods on only partly overlapping sets of genomes, often with inconsistent identifiers and releases—giving right to the cynics’s view that “bioinformatics is ninety percent identifier mapping…”
Enter the Quest for Orthologs consortium
Around that time, Eric Sonnhammer and Albert Vilella organised the first Quest for Orthologs (QfO) meeting at the beautiful Genome Campus in Hinxton, UK—the first of a series of collaborative meetings. We have published detailed reports on these meetings (2009, 2011, 2013; stay tuned for the 2015 meeting report…).
Out of these interactions, the Quest for Orthologs consortium was born, with the mission to benchmark, improve and standardise orthology predictions through collaboration, the use of shared reference datasets, and evaluation of emerging new methods.
The consortium is open to all interested parties and now includes over 60 researchers from 40 institutions worldwide, with representatives from many resources, such as UniProt, Ensembl, NCBI COGs, PANTHER, Inparanoid, PhylomeDB, EggNOG, PLAZA, OrthoDB and our own OMA resource.
The orthology benchmark service and other contributions of the paper
The consortium is organised in working groups. One of them is the benchmarking working group, in which Adrian and I have been very involved. This new paper presents several key outcome of the benchmarking working group.
First and foremost, we present a publicly-available, automated, web-based benchmark service. Accessible at http://orthology.benchmarkservice.org, the service lets method developers evaluate predictions performed on the 2011 QfO reference proteome set of 66 species. Within a few hours after submitting their predictions, they obtain detailed feedback on the performance of their method on various benchmarks compared with other methods. Optionally, they can make the results publicly available.
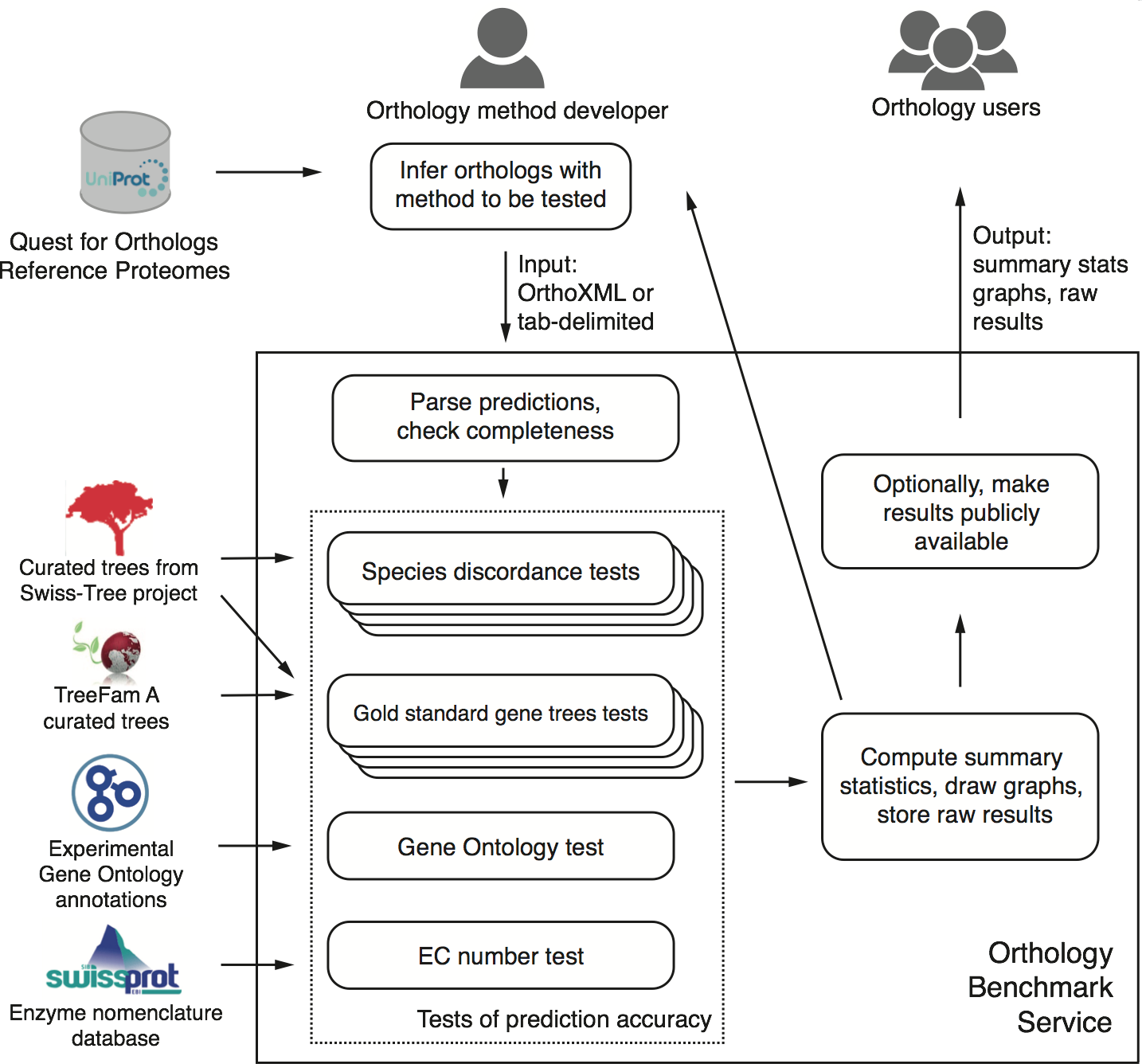
Conceptual overview of the benchmark service (Fig 1 of the paper; click to enlarge)
Second, we discuss the performance of 14 orthology methods on a battery of 20 different tests on a common dataset across all of life.
Third, one of the benchmark, the generalised species discordance test, is new and provides a way for testing pairwise orthology based on trusted species trees of arbitrary size and shape.
Implications
For developers of orthology prediction methods, this work sets minimum standards in orthology benchmarking. Methodological innovations should be reflected in competitive performance in at least a subset of the benchmarks (we recognise that different applications entail different trade-offs). Publication of new or update methods in journals should ideally be accompanied by publication of the associated results in the orthology benchmark service.
For end-users of orthology predictions, the benchmark service provides the most comprehensive survey of methods to date. And because it can process new submissions automatically and continuously, it holds the promise of remaining current and relevant over time. The benchmark service thus enables users to gauge the quality of the orthology calls upon which they depend, and to identify the methods most appropriate to the problem at hand.
References
Altenhoff, A., Boeckmann, B., Capella-Gutierrez, S., Dalquen, D., DeLuca, T., Forslund, K., Huerta-Cepas, J., Linard, B., Pereira, C., Pryszcz, L., Schreiber, F., da Silva, A., Szklarczyk, D., Train, C., Bork, P., Lecompte, O., von Mering, C., Xenarios, I., Sjölander, K., Jensen, L., Martin, M., Muffato, M., Altenhoff, A., Boeckmann, B., Capella-Gutierrez, S., DeLuca, T., Forslund, K., Huerta-Cepas, J., Linard, B., Pereira, C., Pryszcz, L., Schreiber, F., da Silva, A., Szklarczyk, D., Train, C., Lecompte, O., Xenarios, I., Sjölander, K., Martin, M., Muffato, M., Quest for Orthologs consortium, Gabaldón, T., Lewis, S., Thomas, P., Sonnhammer, E., Dessimoz, C., Gabaldón, T., Lewis, S., Thomas, P., Sonnhammer, E., & Dessimoz, C. (2016). Standardized benchmarking in the quest for orthologs Nature Methods DOI: 10.1038/nmeth.3830
Altenhoff, A., & Dessimoz, C. (2009). Phylogenetic and Functional Assessment of Orthologs Inference Projects and Methods PLoS Computational Biology, 5 (1) DOI: 10.1371/journal.pcbi.1000262
Gabaldón, T., Dessimoz, C., Huxley-Jones, J., Vilella, A., Sonnhammer, E., & Lewis, S. (2009). Joining forces in the quest for orthologs Genome Biology, 10 (9) DOI: 10.1186/gb-2009-10-9-403
Dessimoz, C., Gabaldon, T., Roos, D., Sonnhammer, E., Herrero, J., & Quest for Orthologs Consortium (2012). Toward community standards in the quest for orthologs Bioinformatics, 28 (6), 900-904 DOI: 10.1093/bioinformatics/bts050
Sonnhammer, E., Gabaldon, T., Sousa da Silva, A., Martin, M., Robinson-Rechavi, M., Boeckmann, B., Thomas, P., Dessimoz, C., & Quest for Orthologs Consortium. (2014). Big data and other challenges in the quest for orthologs Bioinformatics, 30 (21), 2993-2998 DOI: 10.1093/bioinformatics/btu492
•
Author: David Dylus •
∞
The paper introducing our new tree visualisation tool Phylo.io was just published in MBE.
Yet another tool to display trees, you might say, and indeed, so it is. But for all the tools that have been developed over the years, there are very few that scale to large trees, make it easy to compare trees side-by-side, and simply run in a browser on any computer or mobile device.
To fill this gap, we created Phylo.io.
Story behind the paper
The project started as a student summer internship project, with the aim of producing a tree visualiser that facilitates comparison of trees built on the same set of leaves. After reading the project description, Oscar Robinson, a brilliant student from the Computer Science department at UCL, decided to work on this project during a three month internship. He saw a chance to apply his experience in the development of web tools and to develop his knowledge in the field of data visualisation, one of his major interests.
Once Oscar started with the development of Phylo.io, he realised that only a few tools existed for visual comparison of two trees and either seemed to rely on old technology or were cumbersome to use. Especially this incentive lead him to develop our tool into a fully fledged online resource that is easy to use, platform independent and based on the newest javascript libraries (e.g. D3). Within three months, he managed to produce a prototype of the tool. However, due to the short length of the internship, some details still needed a bit of attention.
Luckily for me, I started my PostDoc in the Dessimoz Lab around that time. Being a novice in a computational lab, Christophe proposed to me to take over the project and bring it to completing as a way to kickstart my postdoc. Altough my computational background at that time did not include any experience in JavaScript programming, I anyway accepted the challenge and was eager to start learning the material. Especially my initial steep learning progress was facilitaed by the help of two other brilliant PhD students, Alex Vesztrocy and Clément Train. Once I acquired some basic understanding, I was able to resolve bugs and add some key missing functionalities such as automatic tree rerooting or persistent storage and sharing functionality.
What is phylo.io and what can it do?
Phylo.io is a web tool that works in any modern browser. All computations are performed client-side and the only restriction on performace is the machine it is running on. Trees can be input in Newick and Extended Newick format. Phylo.io offers many features that other tree viewers have. Branches can be swapped, the rooting can be changed, the thickness, font and other parameters are adaptable. Many of these operations can be performed directly by clicking on a branch or a node in the tree. Importantly, it features an automatic subtree collapsing function: this facilitates the visualisation of large trees and hence the analysis of splits that are deep in the tree.
Next to basic tree visualisation/manipulation it features a compare mode. This mode allows to compare two trees computed using different tools or different models. Similarities and differences are highlighted using a colour scheme directly on the individual branches, making it clear where the differences in two topologies actually are. Additionally, since the output of different tools provides trees with very different rootings and leaf order, Phylo.io has a function to root one of the trees according to the other one and adapt the order of the leaves according to a fixed tree.
How do you use phylo.io?
To save you time, here is a one minute screencast highlighting some of the key features of Phylo.io:
You can find more info in the Phylo.io Manual.
Reference
Robinson, O., Dylus, D., & Dessimoz, C. (2016). Phylo.io: interactive viewing and comparison of large phylogenetic trees on the web
Molecular Biology and Evolution DOI: 10.1093/molbev/msw080