Research
We aspire to better understand gene evolution and function, using statistical and computational methods. The
key questions
underlying our research are:
- How can we best extrapolate our current knowledge of molecular biology,
concentrated in just a handful of model organisms, to the rest of life?
- Conversely, how can we exploit the wealth and diversity of life to better
understand human biology and disease?
- Can we summarize the evolutionary history of species as a sparse mixture of tree topologies?
We tackle these problems by developing statistical and computational
methods and applying them to large-scale genomic data. This process combines
biological aspects in the early stages (e.g. problem statement, identifying
relevant empirical observations, determining dependable benchmarks and
controls), statistical, algorithmic, and computational aspects in the middle
(e.g. model formulation, programming, scaling up), and biological aspects
again at the end in the interpretation of the results.
Representative ongoing projects:
Orthology inference and applications
Orthologs are genes in different organisms that descended from the same
ancestral gene in their last common ancestor. Accurate and comprehensive
identification of these "same genes in different species" is a prerequisite
for numerous biological studies, medical research and pharmaceutical
applications.
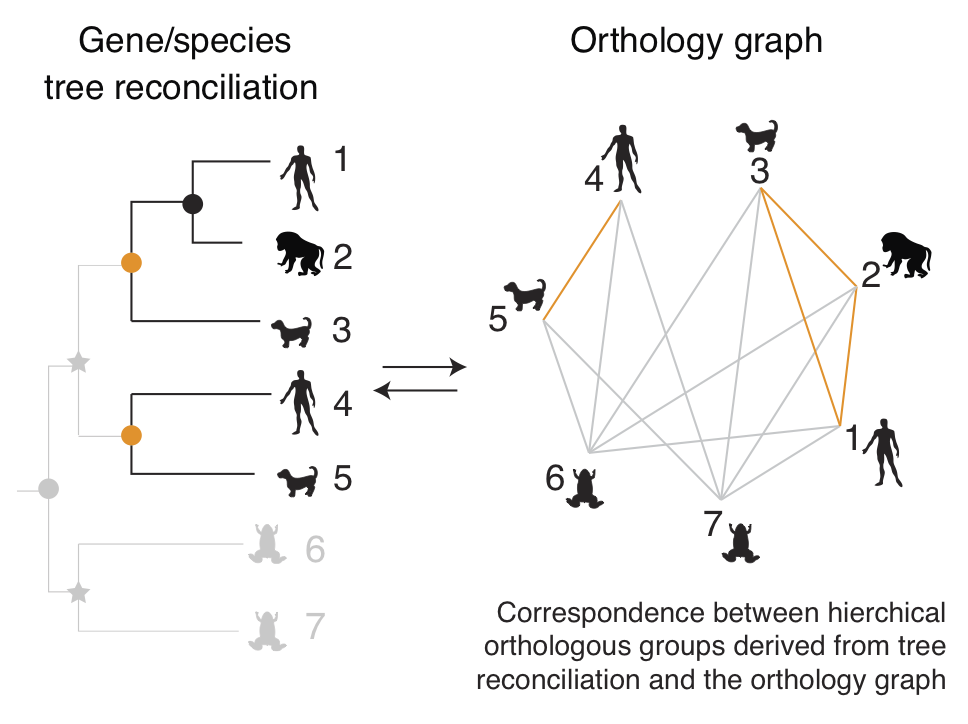
Our main activities in this space are around
OMA (Orthologous MAtrix), an effort to identify orthologous genes among
publicly available genomes. With currently over 2100 genomes analysed, OMA is
among the largest databases of orthologs. Its web interface is consulted hundreds of times
every day, and is linked from several leading sequence databases, including UniProtKB, WormBase, and HGNC.
We are also interested in applications of orthology. For instance, to better
characterise the relationship between gene sequence evolution and gene
function, we tested the validity of the "ortholog conjecture", the notion that
orthologs tend to be functionally more conserved than paralogs.
The story behind this work was written in a
guest post on Jonathan Eisen's blog.
Going the whole HOG (Hierarchical Orthologous Group)
Typical comparative genomics analyses consider either single-copy genes
across multiple species, or multi-copy genes between pairs of species.
Multi-copy genes across multi-species remains hard. Yet genomes are replete
with multi-copy genes.
To overcome this, we have embraced the concept of Hierarchical Orthologous
Groups (HOGs). A HOG comprises all the genes that have descended from a single
gene across a clade of interest. Hence, HOGs relate present-day genes in terms
of their common ancestral genes in key ancestral species. For instance, all
mammalian insulin genes descended from a single ancestral insulin gene at the
root of the mammals; they are thus in one mammalian HOG. But within rodents,
where insulin has duplicated, the two insulin copies in mouse or rat are
placed in distinct rodent HOGs.
The shift from pairwise orthology to HOGs requires new approaches for
inference, benchmarking, visualisation, and integration to downstream
analyses. We are at the forefront of this shift, with several ongoing
projects, e.g. HOG inference and visualisation in the OMA database (funded by
a SIB resource grant), mapping of functional shift onto HOGs (funded by a UK
BBSRC grant), or inference of HOGs in polyploid crop species (research
agreement with BASF CropScience).
Looking forward, we foresee HOGs forming the backbone of any cross-species
comparison—e.g. comparing the evolution of gene splicing across species,
of gene expression, of gene regulation, or indeed of any aspect of gene
function. With the growing importance of non-model species—facilitated by
tools such as CRISPR/Cas9 and sequencing-based assays—multi-species analyses
will further grow in importance.
Big Data Computational Biology
Much of today’s computational biology entails a “Big Data” approach: extracting knowledge from voluminous and
heterogeneous data. This offers new challenges and opportunities. For instance, when we have more data than
we can process, the scalability of our methods becomes more important than their statistical efficiency. A
challenge with very large datasets is that they tempt us to devise ever more complex models, which however
do not necessarily result into better predictions or biological insights (e.g. due to the “curse of
dimensionality” or overfitting). Thus we seek to not only utilise big data when appropriate, but also to
better understand their pitfalls and how of overcome these.
Bio-SODA: Enabling Complex, Semantic Queries to Bioinformatics Databases through Intuitive Searching over
Data (National Research Program 75 Big Data grant with ZHAW and SIB). The goal of this project is to
make it easy for biologists to ask questions integrating knowledge across multiple databases, such as “What
are human-fly orthologs which are expressed in blood and are associated with leukemia?”. We need to solve
three problems: what are the biological entities shared between databases (ontologies), how to query
databases in a decentralised manner, and how to make the system usable by a biologist without specialised
training.
Adaptive approach to sequence clustering (National Research Program 75 Big Data). Current methods for
sequence homology search are either sensitive but slow (all-versus-all alignments), or fast but insensitive
(using k-mer or other approximations, but requiring >80% sequence identity). We aim to searching homologs
using an adaptive, two-layered clustering approach, which is very fast for close variants of known protein
sequences but still sensitive for distant counterparts. This will make it possible to process the huge
datasets produced by metagenomics analyses (which are orders of magnitude larger than UniProt).
Reconstrucing the tree of life: large-scale tree concordance analysis
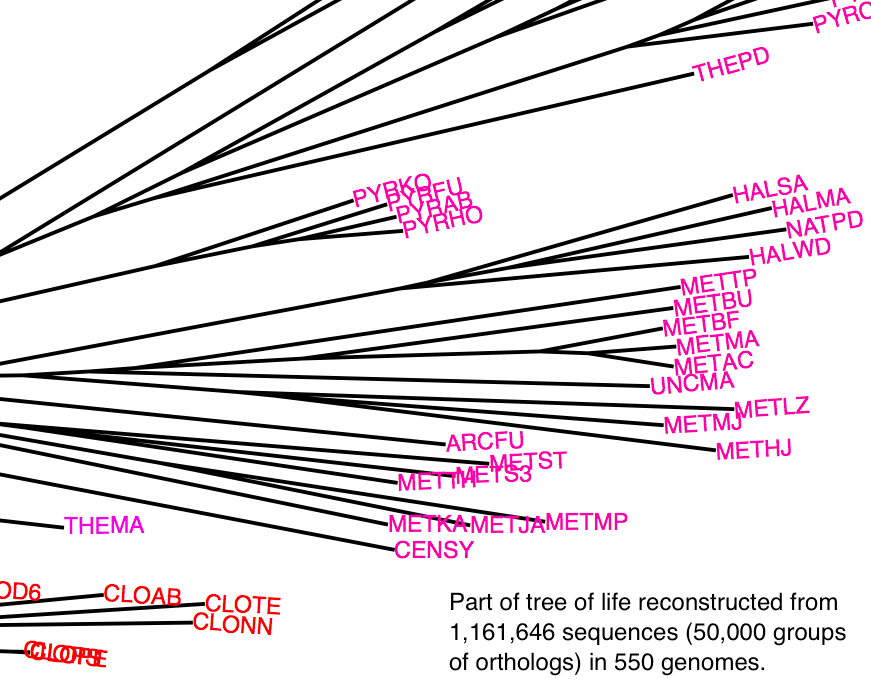
Since Darwin, reconstructing the tree of life has been a major pursuit of
biology. High-throughput genome sequencing is providing us with an abundance
of molecular data, but we still struggle to resolve the deep phylogenies.
Under current methods, adding more characters does not always improve
phylogenetic resolution; and indeed, typical tree reconstruction efforts only
involves a tiny fraction of all genes. In this project, we develop a
phylogenetic tree building method that is sufficiently efficient to take into
account most genes of each species, and that can handle a mixture of
evolutionary histories. Using the majority of genes from a thousand genomes,
we seek to infer the number of different trees that best capture the evolutionary
history of species, to reconstruct these histories, and to visualise them in
an insightful way.
Critical assessment and verification in molecular evolution
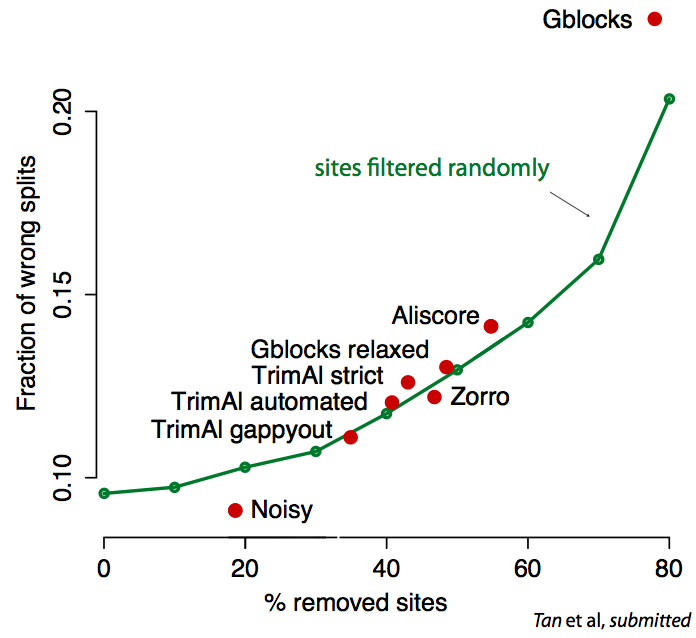
Sequence alignment and phylogenetic tree reconstruction methods are among the
most important contributions of bioinformatics to the life sciences. Both methods
infer past events from current data, be it common ancestry among characters for
alignments, or evolutionary relations among sequences for tree builders.
Because of the inherently unknown nature of these past events,
validation/comparison of the methods (and of their underlying models) is
notoriously difficult. Real data validation is often limited to anecdotal
evidence. In better cases, it consists in some goodness of fit measure (e.g.
AIC). Even then, these measures are based on (implicit) assumptions, which
themselves would need to be tested.
Recently, we have introduced real data tests for orthology inference and for
sequence alignment. Our tests solve the validation problem indirectly—by
assessing the compatibility of a method’s results with general, well-accepted
principles or models. Methods that produce more compatible results are to be
preferred. For instance, the "minimum duplication test" ranks alignment methods
by assuming only that genes evolve along trees, and that the principle of
parsimony applies to gene duplication events.
Quality of computationally inferred gene function annotations
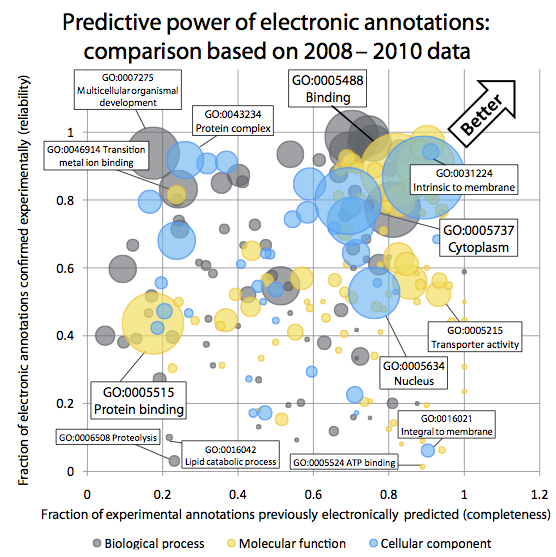 Gene Ontology (GO)
Gene Ontology (GO)
Annotations are a powerful way of capturing the
functional information assigned to gene products. In the Gene Ontology Annotation
database, the largest repository of functional annotations, over 98% of all
function annotations are inferred in silico, without curator oversight. Yet
these "electronic GO annotations" are generally perceived as unreliable and
routinely excluded from analyses. At the same time, we crucially depend on
those automated annotations, as most newly sequenced genomes are non-model
organisms. The key questions we pursue are:
- How can we systematically and quantitatively assess the reliability
of electronic GO annotations?
- Which current inference strategy yields the best predictions?
- In particular, how do evolutionary-based strategies compare with
profile-based strategies?
We have written the story behind some of this work in a
blog post.