•
Author: Kevin Gori •
∞
This is how molecular systematics has worked since the sixties: you take some identifiable feature (e.g. a gene or a protein) common to a group of species and take some measurements of it (e.g. sequencing the DNA). By comparing the results of these measurements you can estimate the evolutionary tree that links the species. Shortly after people started doing this they realised there was a problem: when analyses are based different genes they often estimate different —incongruent — evolutionary trees. As technology has become more capable researchers have begun using more and more genes, so this problem of incongruent trees has moved to the foreground.
There have been lots of good ideas of what do about this problem, and this paper is our contribution. We tried to tackle incongruence by designing a method that groups genes together based on how similar their estimated trees are, without any assumption as to how any incongruence came about.
If all the genes more or less agree on the evolutionary tree, then you get one large group; if some disagree, then they are placed in their own groups. The most interesting case is if several genes disagree in the same way, because then you have an effect to try to explain, and you may have discovered something.
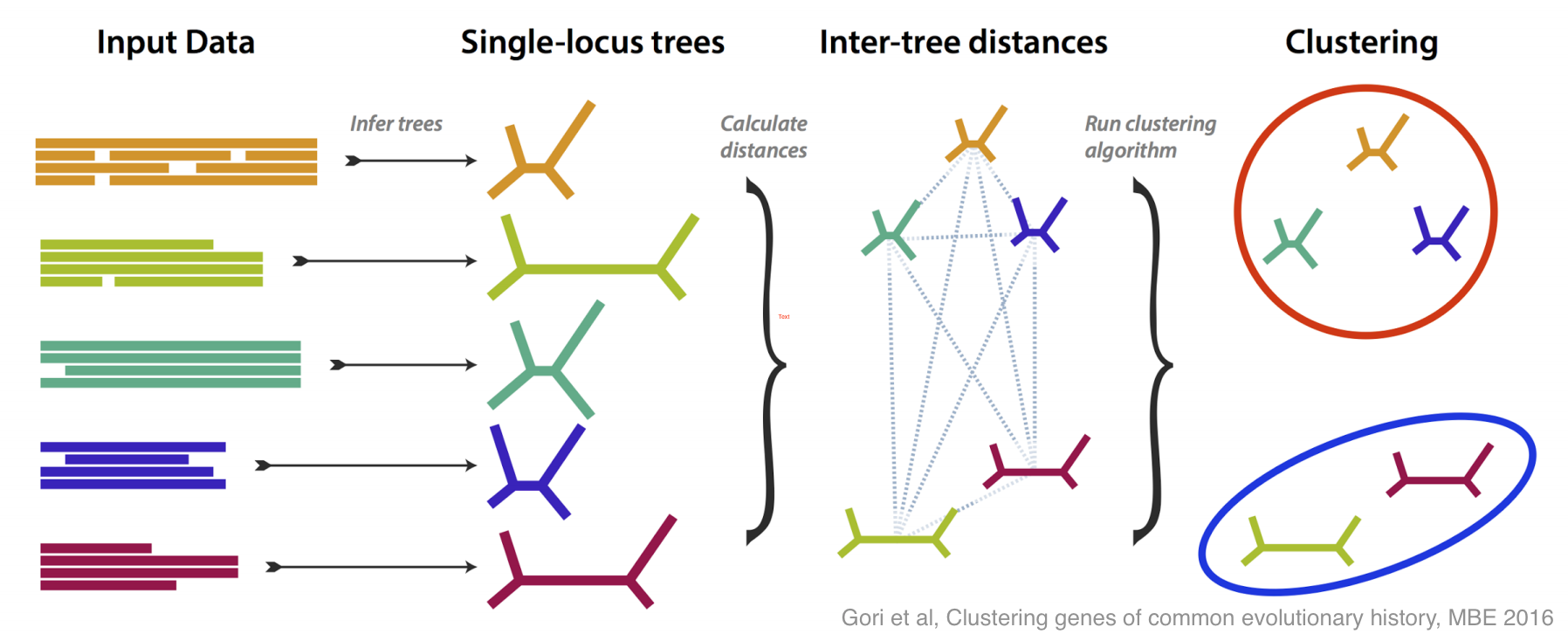
We did lots of simulation to test and refine our method, both in its ability to recognise different incongruent groups, and to estimate how many groups are present. Then, armed with a method that works well on simulation, we tested it on some real data, from yeasts, and from flies.
Our findings were that for the yeast data our method worked really well, and identified 3 distinct groups of genes. The majority of genes were a good fit to the widely accepted tree for the species we looked at. The other two groups showed some major differences, mostly involving two of the species. We had a close look at the data, and concluded that there were some wrong annotations in the data that had introduced sequences that didn’t belong there. This was not the biological result we were looking for, but nonetheless useful.
The flies data were more tricky, as they come from a genus where we aren’t sure how many separate species there are. We produced trees that show better species level resolution than the most recent molecular studies. We also showed high levels of incongruence in the order that the species appear, which can often be the case when species have diverged rapidly, due to a process called incomplete lineage sorting.
So be it to identify artifacts or genuine incongruence among your loci, we think that process-agnostic topology partitioning should become a routine step in phylogenetic analyses. To facilitate this process, we’ve released our code in a new open source software called “treeCl”, available at https://git.io/treeCl.
Reference
Gori K, Suchan T, Alvarez N, Goldman N, & Dessimoz C (2016). Clustering genes of common evolutionary history. Molecular biology and evolution PMID: 26893301