•
Author: Natasha Glover •
∞
Got newly sequenced genomes with protein annotations? Need to quickly and easily define the homologous relationships between the genes?
OMA Standalone is a software developed by our lab which can be used to infer homologs from whole genomes, including orthologs, paralogs, and Hierarchical Orthologous Groups (Altenhoff et al 2019).
The OMA Standalone algorithm works like this:
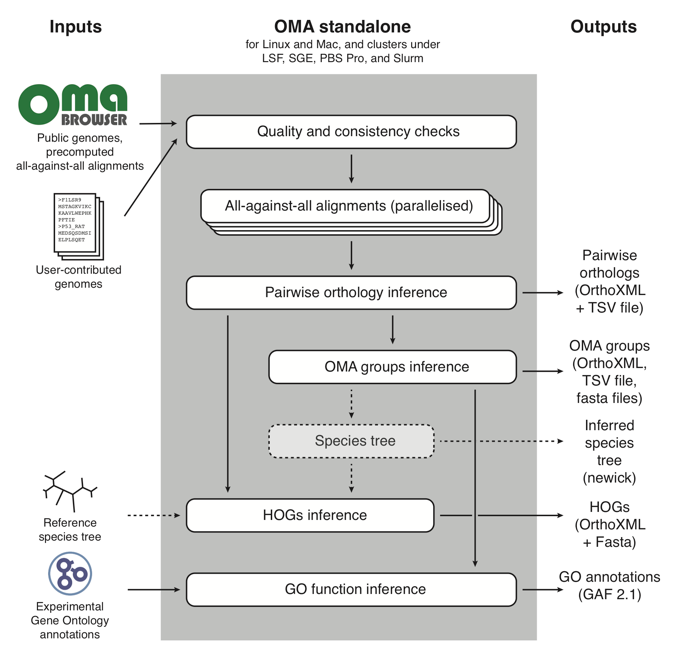
In short, it takes as input user-contributed custom genomes (with the option of combining them with reference genomes already in the OMA database), and proceeds through three main parts:
- Quality and consistency checks of the genomes that will be used to run OMA Standalone;
- All-against-all alignments of every protein sequence to all other protein sequences;
- Orthology inference, in the form of: pairwise orthologs, OMA Groups, and Hierarchical Orthologous Groups (HOGs). For more information on these types of orthologs output by OMA, see OMA: A Primer (Zahn-Zabal et al. 2020).
Although the OMA Standalone is well-documented and straightforward, one of the challenges can be running it on an High Performance Cluster (HPC).
In order to understand the bare necessities needed to run OMA Standalone, we wrote an OMA Standalone Cheat Sheet, which you can download and follow the step-by-step instructions on running the software on an HPC. We use the cluster Wally as an example, as that is one of the HPCs here at the University of Lausanne. Wally uses SLURM as the scheduler for submitting jobs, so all the examples will be shown with that. We plan in the future to provide additional information on running with other schedulers, such as LSF or SGE. In the Cheat Sheet, you will find tips, hints, commands, and example scripts to run OMA Standalone on Wally.
Additionally, we prepared a video which walks the user through the process of running OMA Standalone from start to finish, including:
- Downloading the software
- Preparing your genomes for running
- Editing the necessary parameters file
- Creating the job scripts and
- Submitting your jobs
The video can be found on our lab’s YouTube channel, at OMA standalone: how to efficiently identify orthologs using a cluster, and is also embedded here for your convenience:
We hope these resources can be helpful if you need help getting started running OMA Standalone. But don’t forget, there is also plenty of information that can be found on the OMA Standalone webpage or in the OMA Standalone paper. If all else fails, don’t hesitate to contact us on Biostars.
References
- Altenhoff, A. M. et al. OMA standalone: orthology inference among public and custom genomes and transcriptomes. Genome Res. 29, 1152–1163 (2019).
- Zahn-Zabal, M., Dessimoz, C. & Glover, N. M. Identifying orthologs with OMA: A primer. F1000Res. 9, 27 (2020).