•
Author: Yannis Nevers •
∞
I am excited to introduce our preprint for our new tool OMArk. We hope our software will help fill a gap in assessing the quality of gene annotation sets.
Many studies directly rely on the protein-coding gene repertoires (“proteomes”) predicted from genome assemblies to perform their comparisons. Doing so, they rely on the assumption that the predicted gene content of all genomes are of homogeneous quality and an accurate reflection of reality. Yet in practice, this assumption is rarely met, with protein-coding genes often missing or fragmented in the reported proteomes, non-coding sequences wrongly annotated as coding genes by gene predictors, or contamination from other species wrongly included among the reported sequences.
Why a new proteome quality tool?
Our new method, OMArk, provides a way to easily and comprehensively measure different aspects of proteome quality: completeness of the gene repertoire, consistency of the included genes at the taxonomic level, whether they have doubtful gene structures, and presence or not of inter or intra-domain contamination. Furthermore, contrary to existing methods, OMArk does not rely on a manual selection of reference dataset; instead, it automatically identifies the most likely taxonomic classification of the test proteomes. It can thus process any test proteome across the tree of life using a universal reference database.
Conceptual overview of OMArk (left) and how the innovative consistency assessment is computed (right).
OMArk is accurate and provides new insights
We performed extensive validation of the method by introducing controlled amounts of noise, fragmentation and contamination to reference proteomes and accurately estimating these amounts using OMArk. We also performed a large-scale analysis of 1805 eukaryotic UniProt Reference Proteomes with our software and were able to detect unambiguous cases of quality issues, either caused from incompleteness, contamination, or inclusion of translated non-coding sequences. In the most extreme case, we found a plant proteome with contamination from eight different species—fungi and bacteria.
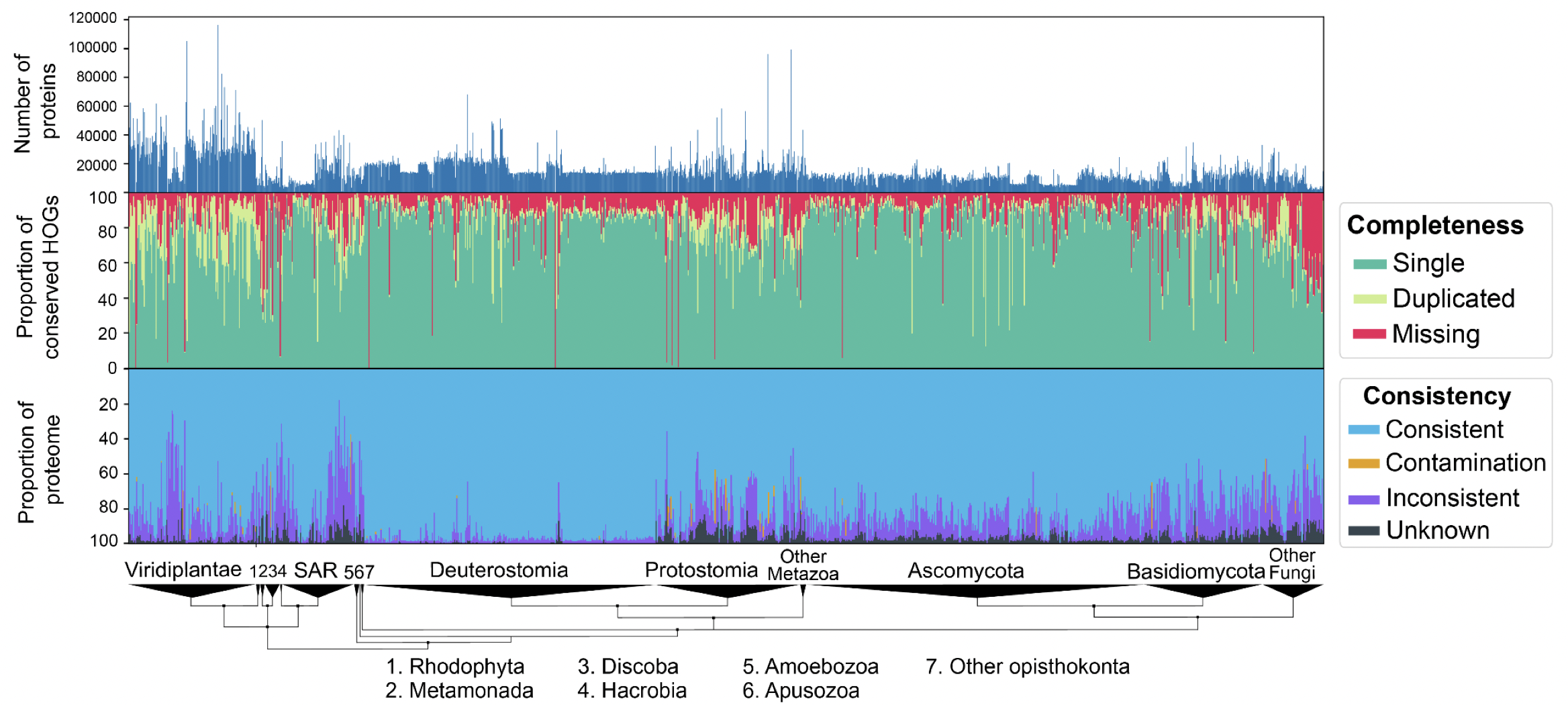
OMArk results on 1805 Eukaryotic proteomes from UniProt. Interactively check results on the OMArk webserver, e.g. for the current cowpea weevil reference proteome.
Why does the consistency metric matter? For example, comparing the Ensembl gene set for two assemblies of Bombus impatiens. We can detect a major improvement in consistency (including contamination removal) for a similar completeness.
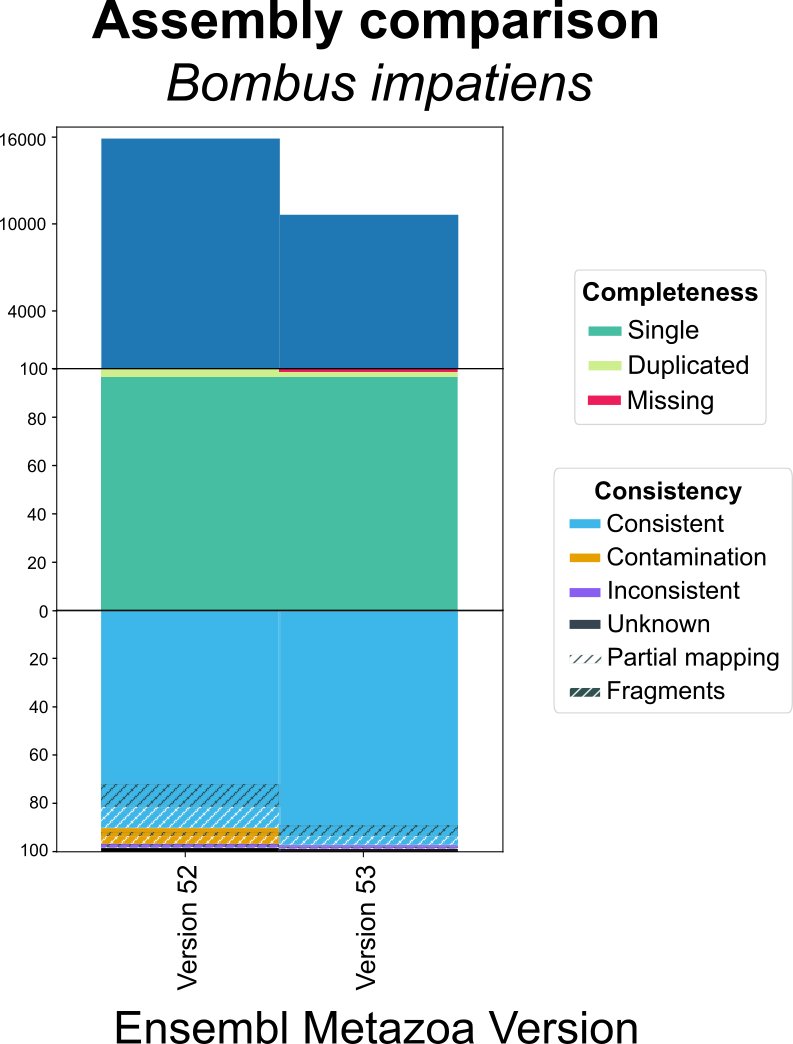
OMArk can reveal improvements in genome assemblies/annotations even if completeness has not substantially changed.
OMArk is quick and easy to run
OMArk can be easily used as a command line tool or on our OMArk webserver. On the webserver, you can submit a FASTA file of your proteome and get results in about 30 minutes. Nothing more required. You can visualize the results and directly compare it to precomputed results from closely related species (UniProt reference proteomes).
More details can be found in the preprint linked below. Please let us know how the tool works for you!
Reference
Yannis Nevers, Alex Warwick Vesztrocy, Victor Rossier, Clément-Marie Train, Adrian Altenhoff, Christophe Dessimoz, Natasha M Glover
Quality assessment of gene repertoire annotations with OMArk
Nature Biotechnology 2024 doi:10.1038/s41587-024-02147-w