•
Author: David Moi •
∞
Breakthroughs don’t come every day, but the consequences of AlphaFold largely solving the 3D structure prediction problem has reshaped biology in profound ways. The sudden availability of protein structures for billions of proteins opens up many new possibilities. Last week’s two papers on the sequencing universe provide a compelling glimpse of the possibilities (here and here).
As someone who has been interested in tracing back the evolutionary origins of selected proteins—such as the cell fusion-mediating proteins fsx1 in plants, viruses, and archaea, or odorant receptors in insects—I have attempted to reconstruct phylogenies from structure in the past.
But I have faced two major issues:
Until AlphaFold came along, there typically wasn’t sufficient high-quality structure predictions as “starting material” to perform structure-based phylogenetics.
Even when I could obtain reasonably high confidence structures, the trees inferred from them were often met with skepticism—how reliable are these trees?
So now that high quality structure predictions are widely available, we could finally ask: are structures any good as starting material to infer trees? Specifically, how accurate are the reconstructed trees compared to sequences?
Today, we are super excited to report that structural phylogenetics works! What’s more, we found an approach that doesn’t just outperform traditional sequence-based methods for distant relationships; it also excels in resolving phylogenetic trees for closely related proteins. This post gives the gist of what we found—the full study is released as a preprint (1).
What’s the big deal with structural phylogenetics?
Before presenting our results, let’s take a step back. Why is structural phylogenetics potentially a big deal? Traditional phylogenetics, the study of evolutionary relationships among species or genes, has long relied on comparing the sequences of DNA, RNA, or proteins. While this approach has been immensely valuable, it does have its limitations. The primary challenge lies in the fact that the sequences of these biomolecules can change rapidly over time due to mutations and other factors, making it difficult to trace back their evolutionary history accurately when the divergence is very high. By contrast, proteins have unique three-dimensional structures that are intricately linked to their functions; these structures tend to change more slowly over evolutionary timescales compared to the sequences of the amino acids that make up the proteins since they are closely tied to the function of the protein.
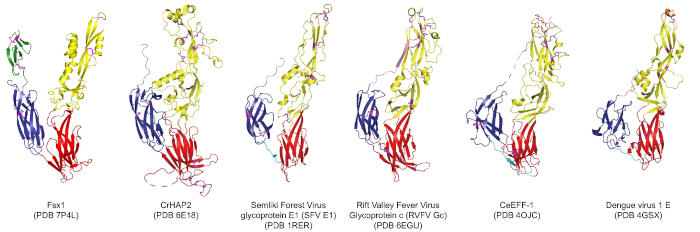
In this particular example close to my heart, we can see structural homology between functionally homologous proteins at wide evolutionary ranges. The examples shown span plants, metazoans, viruses and archaea. They share virtually no sequence homology. Ref: (2)
When we set out to do our work, however, we were not at all sure that it would work, let alone outperform sequence based methods. On the one hand, there have been decades of intensive tool and model refinements for sequence-based approaches, unlike its structure-based counterpart. But also, complications related to structure, such as allostery, flexible regions, and functional constraints could conceivably confound the evolutionary signal that can be extracted from structures.
Evidence that structure-based trees can outperform sequence-based trees
We tested a few structural approaches, and settled on an approach reconstructing distance trees using Foldseek’s “local structural alphabet” approach, which was developed in the lab of our collaborator Martin Steinegger to search for similar structures very rapidly—by encoding local structure motifs in a 20-letter alphabet and repurposing highly optimized alignment software originally developed to align amino acid sequences (3).
Testing and comparing the quality of phylogenetic trees empirically is tricky business. Most comparisons are based on simulated data, or by comparing the fit of data to different models. But how to compare trees that are reconstructed from entirely different kinds of input data? Luckily, our lab has accumulated quite some experience in these kinds of empirical observations, used previously to compare the accuracy of alignment (4 and 5) or orthology (6 and 7) methods. We used an approach which compares the propensity of inferred trees to recapitulate the known taxonomy of the species from which the proteins are sampled from.
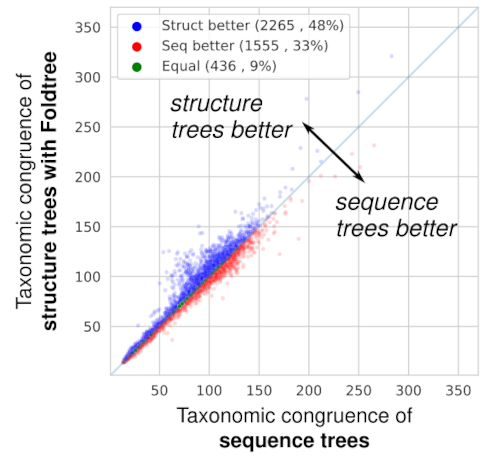
When comparing the taxonomic plausibility of thousands of trees derived from homologous protein families, Foldtree outperforms sequence-based phylogenetics. (In the paper, we show that after filtering the input set to families with high quality structures, the structural phylogenies perform even better!)
Amazingly, the trees we inferred in this way were more in line with the known taxonomy than those defined by sequence similarity! The input data can either be experimental crystal structures or AI structural models. Using good quality structures positively impacts the quality of the trees produced which means that as structural prediction methods get better, so will our structural trees.
The RRNPPA family: a first unifying phylogeny for peptidic quorum sensing proteins
To put our method to the test, we focused on a particularly complex gene family - the RRNPPA quorum sensing receptors (8). These receptors play a pivotal role in enabling communication and coordination among gram-positive bacteria, plasmids, and bacteriophages for crucial behaviors like sporulation, virulence, antibiotic resistance, conjugation, and phage lysis/lysogeny decisions.
The complex evolutionary pattern of this family is revealed in its name. Before AI structures, new homologs were previously only detectable after having been crystallized and each subfamily was added piecemeal to the overall picture, resulting in their particularly long acronym. As the family expanded researchers also attempted to piece together its evolutionary history, using a diverse set of methods, some of which relied on structural analysis. Using Foldtree we decoded the evolutionary diversification of these genes, shedding new light on their intricate history.
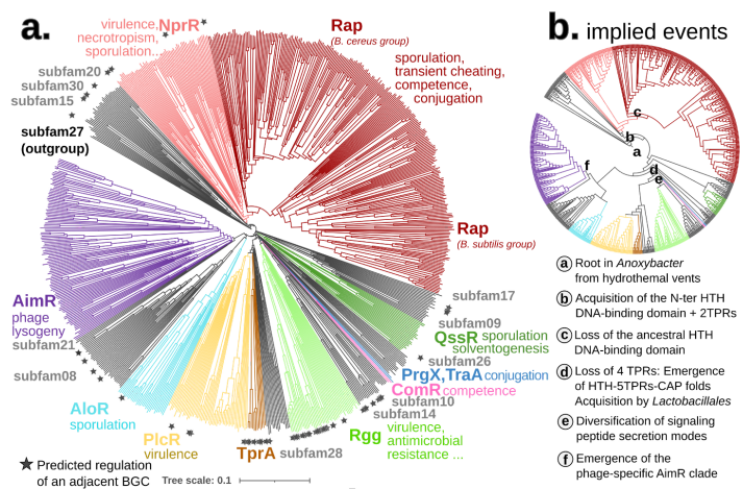
Compared to the sequence-based phylogeny, the Foldtree reconstruction of the RRNPPA family’s history is remarkably parsimonious. Several events such as domain architecture changes or transfers to the viral world appear only once in the tree.
Foldtree: infer a structural phylogeny for your favorite protein family

To make it easy to try this approach, as well as facilitate methodological improvements, we are releasing this new approach as an open source tool we call Foldtree. It’s available for download on GitHub (https://github.com/DessimozLab/fold_tree). Try it on your favorite protein family and let us know how it performs!
Exciting new research directions
High-accuracy structural phylogenetics has the potential to uncover deeper evolutionary relationships, elucidate unknown protein functions, and even refine the design of bioengineered molecules. The evolutionary histories of protein families in the viral domain, the start of eukaryotic life and the role of asgard archaea as well as the evolution of the prokaryotic mobilome are just a few cases where the fast pace of evolution has confounded sequence-based analyses and could be revisited. We believe this work represents an important step in investigating how structures are polished by the processes of evolution and how we can use this signal to peer further into the past than ever before.
References
Moi D, Bernard C, Steinegger M, Nevers Y, Langleib M, Dessimoz C. Structural phylogenetics unravels the evolutionary diversification of communication systems in gram-positive bacteria and their viruses. bioRxiv 2023.09.19.558401; doi: https://doi.org/10.1101/2023.09.19.558401
Moi D, Nishio S, Li X, Valansi C, Langleib M, Brukman NG, et al. Discovery of archaeal fusexins homologous to eukaryotic HAP2/GCS1 gamete fusion proteins. Nat Commun. 2022;13: 3880. doi:10.1038/s41467-022-31564-1
van Kempen M, Kim SS, Tumescheit C, Mirdita M, Lee J, Gilchrist CLM, et al. Fast and accurate protein structure search with Foldseek. Nat Biotechnol. 2023. doi:10.1038/s41587-023-01773-0
Tan G, Gil M, Löytynoja AP, Goldman N, Dessimoz C. Simple chained guide trees give poorer multiple sequence alignments than inferred trees in simulation and phylogenetic benchmarks. Proceedings of the National Academy of Sciences of the United States of America. 2015. pp. E99–100. doi:10.1073/pnas.1417526112
Dessimoz C, Gil M. Phylogenetic assessment of alignments reveals neglected tree signal in gaps. Genome Biol. 2010;11: R37. doi:10.1186/gb-2010-11-4-r37
Altenhoff AM, Dessimoz C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput Biol. 2009;5: e1000262. doi:10.1371/journal.pcbi.1000262
Altenhoff AM, Boeckmann B, Capella-Gutierrez S, Dalquen DA, DeLuca T, Forslund K, et al. Standardized benchmarking in the quest for orthologs. Nat Methods. 2016;13: 425–430. doi:10.1038/nmeth.3830
Bernard C, Li Y, Lopez P, Bapteste E. Large-scale identification of known and novel RRNPP quorum sensing systems by RRNPP_detector captures novel features of bacterial, plasmidic and viral co-evolution. Mol Biol Evol. 2023. doi:10.1093/molbev/msad062