•
Author: Nastassia Gobet •
∞
When I started using word processors, the spell checker was only looking at small and common typing errors and was often trying to correct acceptable words due to lack of vocabulary. A few years later, they not only are better at it and use more developed dictionaries, but they can also capture grammar mistakes and redundant phrases. A similar story is happening with the detection of genomic variants.
The genome as a big text
The genome can be considered as a big text, written in a 4-letter alphabet (A, C, G, T). When comparing the genomic words from two individuals, we can look at single or few letter(s) differences (single nucleotide variants, SNVs) and longer patterns (structural variants, SVs) such as words, sentences, and paragraphs that are added (insertions) or missing (deletions), exchanged (translocations), repeated (duplications and copy number variations, CNVs), inverted (inversions) or combinations of these (complex SVs).
Discovering the importance of SVs
About ten years ago, the focus was mainly on SNVs as these are numerous and many methods to detect them were developed. They were studied in deep and indexed in dictionaries (databases) that also document their frequencies. However, one letter differences do not necessarily have a significant effect on the meaning of the text (the phenotypes). On the other hand, although SVs were underestimated and consequently understudied, they were discovered to have a profound phenotypic impact on gene regulation, dosage, and function. Therefore, they are important in a wide variety of medical conditions: cancers, neurological diseases (Parkinson, Huntington), and mental disorders (autism, schizophrenia).
Challenges in SV identification
Methods were recently developed and are currently being developed to detect SVs. A number of challenges need to be dealt with. First, short read sequencing greatly limits the detection of large events exceeding read length. Consequently, using longer read technologies (PacBio and ONT) is improving the range of detectable SVs, but this comes at the cost of decreased sequencing accuracy and higher price. Hybrid strategies combining short and long reads are therefore promising. Second, SVs are hard to classify as the variant type depends on variant sequence context: a sequence can be considered an insertion, duplication, or translocation depending on the source (Figure 1). In addition, the number of possible SVs is infinite, whereas for SNVs there are 3 variants per position in the worst case. SVs are thus hard to compare: which criteria should we use to determine if two slightly different calls correspond to the same event or not? This affects SV reporting and frequencies. Due to the relative youth of the field, standards and best practices have yet to be established. Different initiatives (eg. Genome in a Bottle and SEQC2) aim at better characterizing false positives and false negatives in SV calling. This should help implement more objective benchmarking and comparison between the various detection methods.
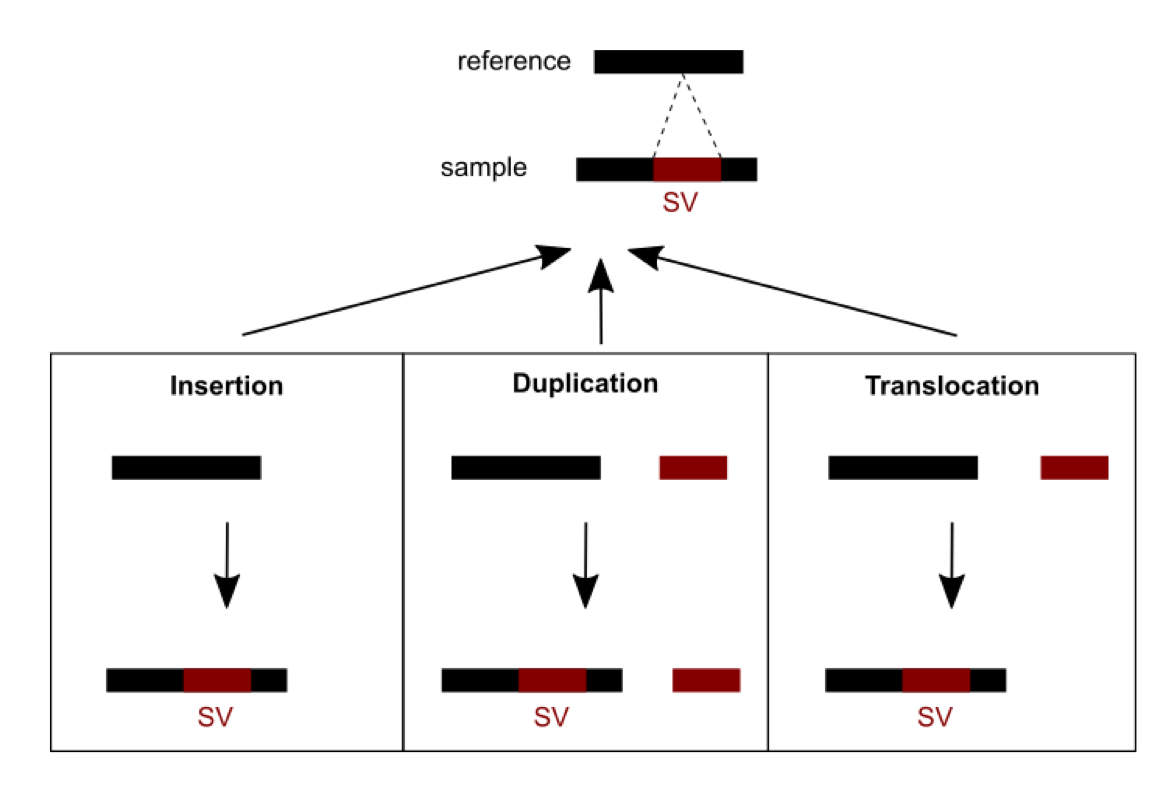
Figure 1: An SV was called for a sequence from a sample differing from the reference sequence. Three possible scenarios of formation could explain the SV observed: an insertion, a duplication or a translocation.
Future of genomic spelling and grammar checkers
Standards and objective benchmarking for SV detection are still missing, so one must be careful with results obtained from current methods. However, SVs are increasingly recognized as being important and technologies to detect them are evolving rapidly. I think their use will become a more common practice in genomic variation studies in a few years, similar to spelling and grammar checkers in text processors. And you, which genome checker will you use?
Reference
Mahmoud M, Gobet N, Cruz-Dávalos DI, Mounier N, Dessimoz C, Sedlazeck FJ. 2019. Structural variant calling: the long and the short of it. Genome Biol 20:246. doi:10.1186/s13059-019-1828-7.
If you want to get involved in improving SV variant detection, consider joining this Hackathon, to be hold remotely Oct. 11-14, 2020.