•
Author: Charles Bernard •
∞
For an evolutionary biologist, tracing today’s genomes back to key ancestors on the Tree of Life is a dream come true. With a collection of ancestral genomes, we could unravel the genetic steps that led to Life’s diversification from LUCA, the Last Universal Common Ancestor.
In practice, this means comparing modern genomes to find similar features—“orthologous” genes—passed down from common ancestors. By reversing this thinking, we can use these orthologous features as clues to “reconstruct” what ancestral genomes might have looked like.
But while much previous work has focused on reconstructing ancestral gene repertoires, reconstructing ancestral gene orders has been much more elusive.
In this post, I’ll dive into how we’ve developed a tool, EdgeHOG (1), to achieve this at a scale and speed never seen before (preprint here: https://www.biorxiv.org/content/10.1101/2024.08.28.610045v1).
Why do ancestral gene orders matter?
A genome isn’t just a random collection of genes; it has a structure that’s been shaped by evolution. Where a gene sits on a chromosome and its neighbours can matter a lot. Indeed, neighboring genes often work together (2). Plus, changes in gene order—genomic rearrangements—can lead to new traits and adaptations (3).
So, to understand the evolutionary history of these gene neighborhoods, we need to focus on gene adjacencies, not just the genes themselves. However, figuring out the gene order for every internal node on the Tree of Life is a huge computational challenge (4)…
How did we get into ancestral gene order inference?
When we wanted to analyze the link between gene function and gene adjacency across Life, we needed software that could reconstruct ancestral gene orders across the entire Tree of Life in one go, while accurately distinguishing between different copies of a gene in an ancestor.
But no tool could scale up to this level. Even the best tools, like AGORA (5) require reconstructing gene trees and perform pairwise comparisons of gene orders, which make them too slow to run on large datasets.
This is what drove us to create an algorithm with a clear goal: a linear time approach to reconstruct ancestral gene order, but without sacrificing accuracy.
How does EdgeHOG achieve linear-time complexity?
To infer ancestral gene orders at scale, our approach uses Hierarchical Orthologous Groups (HOGs). These model the lineage of genes from their ancestors to today’s species, assuming vertical inheritance.
By leveraging these gene lineages, our method uses “tree traversal” tricks to propagate or remove gene adjacencies along the species tree without any pairwise comparisons. Thanks to these tricks, our approach scales linearly with the size of the input phylogeny.
Since the software draws edges (gene adjacencies) between HOGs (proxies for ancestral genes), we called it EdgeHOG.
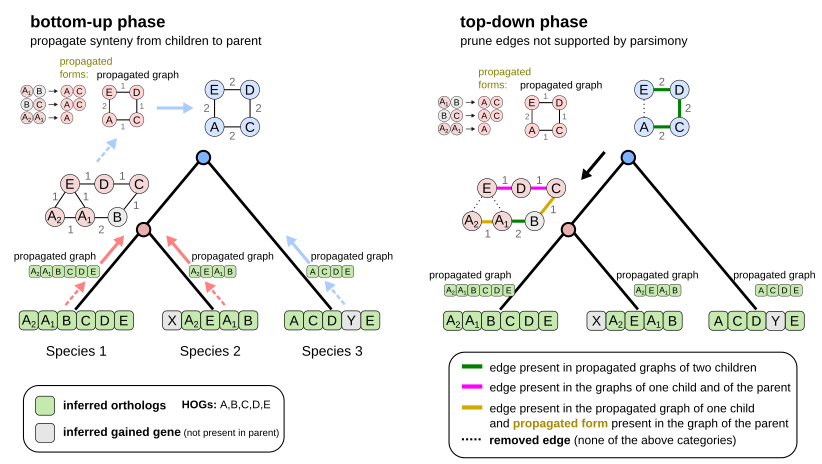
Here are the 2 first steps of EdgeHOG and the famous tree traversal tricks! The bottom-up phase propagates gene adjacencies up to the parental level of the species tree as long it is inferred by the HOG framework to have the two ancestral genes. The top-down phase essentially applies the Fitch algoritm and removes edges not supported by parsimony. Designing these tricks to comply with the constraint of linear time complexity was probably the most fun part of the project!
Fast and accurate
But EdgeHOG is not only fast, it is also very accurate! We validated it extensively on both simulated and real data. Across all benchmarks, EdgeHOG’s precision and recall met or exceeded the state of the art.
How to access EdgeHOG’s large scale inference of ancestral genomes?
The next step for us was to apply EdgeHOG to the entire OMA database, which currently includes 2,845 genomes from across the Tree of Life! This represent the first tree-of-life scale inference, resulting in 1133 ancestral genomes. You can explore these genomes on the OMA browser by clicking on Explore → Quick access to → Extant and ancestral genomes. For instance, check out the ancestral gene order for the last common ancestor of the mammals.
In the EdgeHOG paper, we also analysed the functions of the first ancestral contigs of genes ever reconstructed for LECA, the Last Eukaryotic Common Ancestor! These contigs contain genes that highlight core pathways like glycolysis, the pentose-phosphate shunt, amino-acid recycling, and histone organisation.
What kind of evolutionary analyses does EdgeHOG unlock?
In the lab, we’re using EdgeHOG to study the association between between gene order conservation and function conservation across different branches of the Tree of Life. We’re also dating gene adjacencies to identify old genomic neighbourhoods (like histone clusters in eukaryotes) or newer ones (like gene adjacencies on the sex chromosomes of animals).
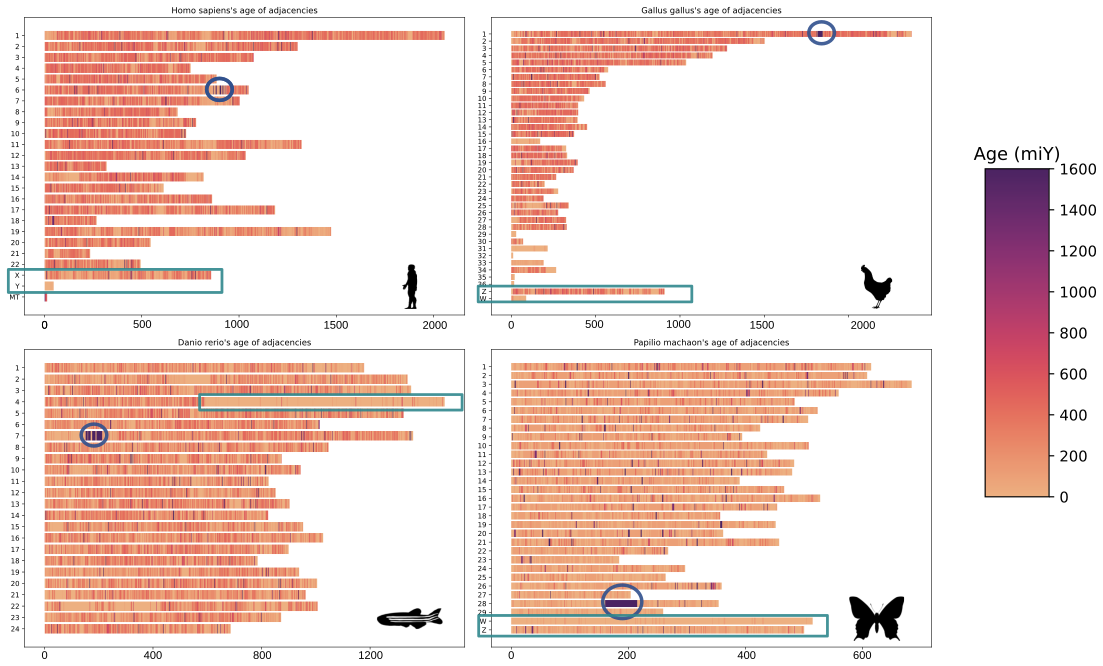
On these karyotypes, old histone clusters are circled in blue, and sex chromosomes are highlighted by rectangles. The estimated age of adjacencies is shown by the color scale on the right.
Overall, EdgeHOG opens up new possibilities in comparative genomics. For example, it helps track genomic rearrangements across a species tree, identify conserved gene clusters in clades of interest, or improve genome assembly by integrating gene order data from other species. Ultimately, knowing ancestral gene orders will enhance orthology inference by spotting highly divergent orthologs through their neighboring genes.
Do you want to try EdgeHOG on your datasets?
EdgeHOG is easy to use and available on GitHub https://github.com/DessimozLab/edgehog.
You’ll need a species tree, proteomes of each extant species (in Fasta files), and gene coordinates on contigs (in GFF files). Then, run our superfast FastOMA method to infer the HOGs. Finally, call EdgeHOG with the HOGs (OrthoXML file), the species tree (Newick file), and the path to the GFF files.
Now, you’re ready to perform big data ancestral gene order inferences, even with massive phylogenies of over 1,000 species! Try it on your favorite clade and let us know how it goes!
References
Bernard C, Nevers Y, Karampudi NBR, Gilbert KJ, Train C, Warwick Vesztrocy A, Glover N, Altenhoff A, Dessimoz C. EdgeHOG: fine-grained ancestral gene order inference at tree-of-life scale. bioRxiv 2024. https://doi.org/10.1101/2024.08.28.610045
Overbeek R, Fonstein M, D’Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci U S A. 1999. https://doi.org/10.1073/pnas.96.6.2896
An X, Mao L, Wang Y, Xu Q, Liu X, Zhang S, Qiao Z, Li B, Li F, Kuang Z, Wan N, Liang X, Duan Q, Feng Z, Yang X, Liu S, Nevo E, Liu J, Storz JF, Li K. Genomic structural variation is associated with hypoxia adaptation in high-altitude zokors. Nat Ecol Evol. 2024. https://doi.org/10.1038/s41559-023-02275-7
El-Mabrouk N. Predicting the Evolution of Syntenies—An Algorithmic Review. Algorithms. 2021. https://doi.org/10.3390/a14050152
Muffato M, Louis A, Nguyen NTT, Lucas J, Berthelot C, Roest Crollius H. Reconstruction of hundreds of reference ancestral genomes across the eukaryotic kingdom. Nat Ecol Evol. 2023. https://doi.org/10.1038/s41559-022-01956-z