•
Author: Kevin Gori •
∞
This is how molecular systematics has worked since the sixties: you take some identifiable feature (e.g. a gene or a protein) common to a group of species and take some measurements of it (e.g. sequencing the DNA). By comparing the results of these measurements you can estimate the evolutionary tree that links the species. Shortly after people started doing this they realised there was a problem: when analyses are based different genes they often estimate different —incongruent — evolutionary trees. As technology has become more capable researchers have begun using more and more genes, so this problem of incongruent trees has moved to the foreground.
There have been lots of good ideas of what do about this problem, and this paper is our contribution. We tried to tackle incongruence by designing a method that groups genes together based on how similar their estimated trees are, without any assumption as to how any incongruence came about.
If all the genes more or less agree on the evolutionary tree, then you get one large group; if some disagree, then they are placed in their own groups. The most interesting case is if several genes disagree in the same way, because then you have an effect to try to explain, and you may have discovered something.
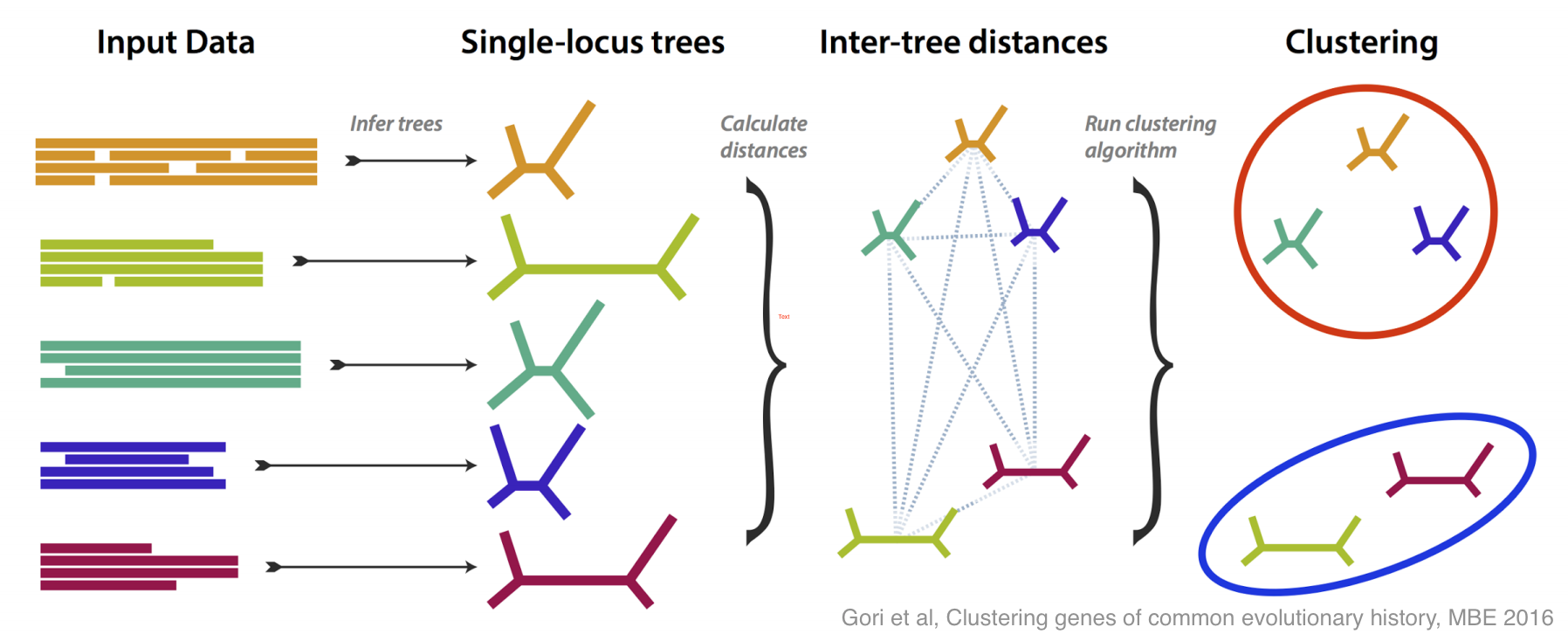
We did lots of simulation to test and refine our method, both in its ability to recognise different incongruent groups, and to estimate how many groups are present. Then, armed with a method that works well on simulation, we tested it on some real data, from yeasts, and from flies.
Our findings were that for the yeast data our method worked really well, and identified 3 distinct groups of genes. The majority of genes were a good fit to the widely accepted tree for the species we looked at. The other two groups showed some major differences, mostly involving two of the species. We had a close look at the data, and concluded that there were some wrong annotations in the data that had introduced sequences that didn’t belong there. This was not the biological result we were looking for, but nonetheless useful.
The flies data were more tricky, as they come from a genus where we aren’t sure how many separate species there are. We produced trees that show better species level resolution than the most recent molecular studies. We also showed high levels of incongruence in the order that the species appear, which can often be the case when species have diverged rapidly, due to a process called incomplete lineage sorting.
So be it to identify artifacts or genuine incongruence among your loci, we think that process-agnostic topology partitioning should become a routine step in phylogenetic analyses. To facilitate this process, we’ve released our code in a new open source software called “treeCl”, available at https://git.io/treeCl.
Reference
Gori K, Suchan T, Alvarez N, Goldman N, & Dessimoz C (2016). Clustering genes of common evolutionary history. Molecular biology and evolution PMID: 26893301
•
Author: Natasha Glover •
∞
We know homologs are genes related by common ancestry. But throw complex evolutionary events into the mix and things can get little dicey. Under the umbrella of homologs exist many different categories: orthologs, paralogs, ohnologs, xenologs, co-ortholog, in-paralogs, out-paralogs, paleologs, among others. All of these —log terms have a specific meaning (see my previous blog post on orthology and paralogy), but now we will focus on one in particular: homoeologs.
But before we get into the definition, let’s start at the beginning. When I started as a postdoc at Bayer CropScience working with Henning Redestig in collaboration with Christophe Dessimoz University College London, I was tasked with evaluating homoeolog predictions using the OMA algorithm.
What are homoeologs?
From my previous experience, I knew homoeologs as roughly “corresponding” genes between subgenomes of a polyploid organism. For example, the wheat genome is an allohexaploid, with 3 diploid subgenomes named A, B, and D. Given a gene on chromosome 3B, you will most likely find a nearly identical copy on chromosomes 3A and 3D, in roughly the same position. These corresponding copies across subgenomes are known as homoeologs. But this definition left something to be desired— it didn’t tell me anything about the evolutionary relationship between the homoeologs. Worse, it was ambiguous in that it required discretionary similarity thresholds in terms of sequence and positional conservation. How could we test for performance if there was no unambiguous definition of the target?
Time to hit the books
Like many researchers starting a new project, I went to the scientific literature to get more information. After many hours spent on google scholar, I found myself with more questions than answers. Firstly, what were the evolutionary events that give rise to homoeologs? How do they fit in with the other —log terms? Can they be found only in a certain type of polyploid, but not another? How do things like gene duplication and movement affect our understanding of what a homoeolog is? And finally, after seeing it the word written as homoeolog, homeolog, and homoeologue, how do you even spell it?
There are some excellent review papers out there on polyploidy which shed light on the biological consequences of homoeology. This, this, or this for example. However, when searching the whole of the literature, I found many inconsistent, vague, or even incorrect usages of the term homoeolog. Sometimes people defined homoeologs on the basis of their chromosome pairing patterns. Other times homoeologs were used to describe corresponding genes from different, although closely related species. Many papers said homoeologs were necessarily syntenic. Others don’t define the term at all.
Getting on the same page
These imprecise or incorrect definitions can lead to confusion. In recent years, advances in technology has afforded us the opportunity to sequence many new genomes, including polyploids. All these new techniques and have exploded the amount of data and brought about collaborations between geneticists, molecular biologists, plant breeders, bioinformaticians, phylogeneticists, and statisticians. Therefore we think it’s important to have a precise and evolutionary meaningful definition of homoeology as a reference point.
What we learned
Thus we went back to the earliest usage of the term we could find and synthesizing the literature to date. We define homoeologs as “pairs of genes or chromosomes in the same species that originated by speciation and were brought back together in the same genome by allopolyploidization”. For recent hybrids, as long as there was no rearrangement across subgenomes, homoeologs can be thought of as orthologs between these subgenomes. Here’s how they fit in with other common homologs:
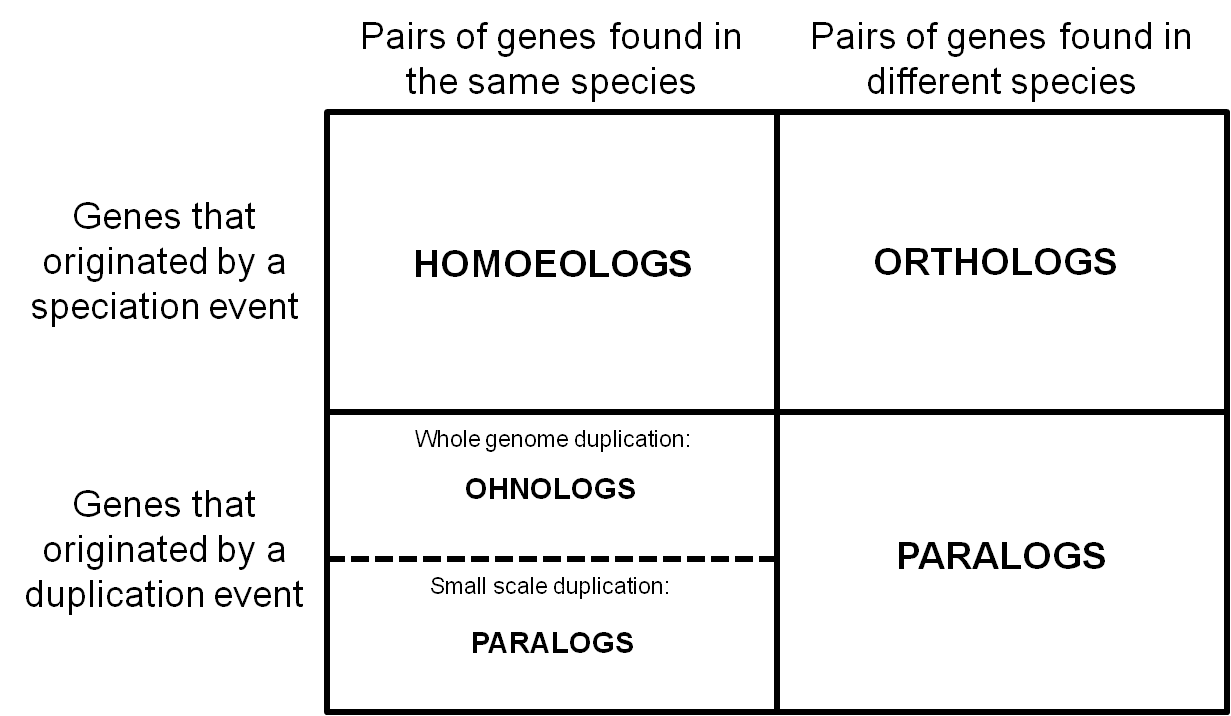
We realized that homoeologs are not necessarily one-to-one or syntenic. Depending on the particular patterns of gene duplication and rearrangement in a given species, we may see homoeologs at a 1:many or across non-corresponding chromosomes.
We also reviewed homoeolog inference techniques, starting from low-throughput lab techniques to evolution-based computational methods. Orthology prediction is a booming area of active research, so many orthology inference methods can be applied to homoeology prediction.
Last but not least, we learned that even though homoeolog has alternatively been spelled “homeolog” (no extra o), homoeolog is the clear winner in terms of popularity. The “homoeo—” spelling has been used more than double the amount of times in the literature. Fortunately however, both are pronounced the same (“ho-mee-o-log”)
Check out the review paper in Trends in Plant Science (open access!). We hope this paper can serve as a jump off point for those interested in tackling homoeology, especially for those new to the field.
Reference
Glover, N., Redestig, H., & Dessimoz, C. (2016). Homoeologs: What Are They and How Do We Infer Them? Trends in Plant Science DOI: 10.1016/j.tplants.2016.02.005
•
Author: Christophe Dessimoz •
∞
A few months ago, we published a paper that spent four years in peer-review (story behind the paper). Because of this, I feel entitled to an opinion on the pre- vs post-publication review debate.
Background on preprints and their effect on peer-review
If you have been living under a rock, or if you are not on Twitter, you may not have noticed that preprints are becoming more widely accepted in biology—supported by initiatives such as Haldane’s Sieve and bioRxiv. This is particularly true in population genetics, evolutionary biology, bioinformatics, and genomics. Typically, a manuscript is made available as preprint just as it is submitted to a scientific journal, and therefore prior to peer-review. I am saying “made available” instead of “published” because although preprints can be read by anybody, the general view is that the canonical publication event lies with the journal, post peer-review. Because of this, many traditional journals tolerate this practice: peer-review technically remains “pre-publication” and the journals get to keep their gatekeeping function.
The key benefit of preprints is that they accelerate scientific communication. Indeed, peer-review can be long and frustrating for authors. Reviewers sometimes misjudge the importance of papers or request unreasonable amounts of additional work. The ability to bypass peer-review can thus be liberating for authors. Thus, if we instead recognised preprints as the canonical publication event, so goes the idea, peer-review would be relegated to a secondary role and journals would loose their gatekeeping function. This is the “post-publication” peer-review model.
For more background info, here are a few pointers:
Advantages of pre- and post-publication peer-review
What did our recent experience teach us? Spending four years in various stage of peer-review is a huge strain on the authors, reviewers, and editors. On the positive side, the final paper was more complete (some of the methods tested were published after our first submission!). Undoubtedly, it became a clearer and more solid paper. However, as I pointed out in my post on the paper, our main conclusions did not change. They could have been brought to everyone’s attention four years earlier.
So should pre-publication peer-review be abolished? In this particular case, it’s debatable. If we had known what awaited us, we would have released the manuscript as a preprint (eg on arXiv, bioRxiv, or PeerJ PrePrint)—something we have done with subsequent pieces of work.
However in general, I still think that pre-publication peer-review has many merits. First, thankfully this experience was extreme; on average things are much faster: 2-4 months including one revision cycle is quite typical in my experience. With some journals, this can be even faster (Bioinformatics, PeerJ or MBE jump to mind). Second, pre-publication peer-review can identify flaws or interesting points overlooked by the authors—to the point that in some cases (large multi-author studies!), peer-reviewers wind up contributing almost certainly more to the paper than some of the co-authors. Furthermore, while reviewers do not always agree in their comments, when they do, the authors better pay attention.
That being said, unorthodox or controversial results can be extremely difficult to publish under the pre-publication peer-review model, particularly if some of the reviewers have vested interests in the status quo.
Best practices at the age of pre- and post-publication peer-review
So what model am I arguing for? I think the emerging combination of preprints and journals can give us the best of the two worlds. Preprints ensure that advances can be quickly, broadly, and unimpededly disseminated. Journals add a layer of quality control and differentiation, and even glamour if so they choose. Importantly, this new paradigm shifts power from the publishers back to us, the researchers. And you all remember what comes with great powers, right?
As peer-reviewers, it is our job to identify specific issues with the work, and bring them to the authors and the editor, but ultimately, we should remember that the work we review is not our work. If the authors choose to ignore points we consider important, it may be more constructive and rewarding to write a rebuttal paper anyway. Post-publication peer-review as it were!
As editors, we should pay attention to potential conflicts of interests, focus on a limited set of key points that need addressing, and remember that every additional round of revisions costs precious time and resources. The additional delay could result in wasteful duplication of the work by others, or missed opportunities to build upon the findings. Thus we have a moral obligation to balance pre- and post-publication peer-review. Too often, editors lazily or cowardly repeatedly forward all reviewer comments back and forth without taking a stance, with little consideration of the burden this incurs to the authors and the rest of the community.
As authors, one simple but powerful thing we can do is to more openly acknowledge the shortcomings of our work and candidly disclose unresolved issues. In case of fundamental disagreeement with a peer-reviewer, the impasse may be overcome by including an account of the disagrement as part of the paper. In fact, this is precisely what we ended up doing in our paper. In the discussion section, we wrote:
And sixth, we disclose that in spite of the several lines of evidence and
numerous controls provided in this study, one anonymous referee remained
skeptical of our conclusions. His/her arguments were: (i) instead of using
default parameters or globally optimized ones, filtering parameters should
be adjusted for each data set; (ii) the observations that, in some cases,
phylogenies reconstructed using a least-squares distance method were more
accurate than phylogenies reconstructed using a maximum likelihood method
(Supplementary Figs. 7–10 available on Dryad at
http://dx.doi.org/10.5061/dryad.pc5j0), and that ClustalW performed
“surprisingly well” compared with other aligners, are indicative that the
data sets used for the species discordance test are flawed; (iii) the
parsimony criterion underlying the minimum duplication test and the Ensembl
analyses is questionable.
Indeed, not every issue can be resolved during peer-review. At some point, the debate should happen in the open. Any one single paper is rarely the “last word” on a question anyway. And as our editor admitted, a bit of controversy is good for the journal.
Reference
Tan G, Muffato M, Ledergerber C, Herrero J, Goldman N, Gil M, & Dessimoz C (2015). Current Methods for Automated Filtering of Multiple Sequence Alignments Frequently Worsen Single-Gene Phylogenetic Inference. Systematic biology, 64 (5), 778-91 PMID: 26031838
 The Dessimoz Lab
blog is licensed under a Creative
Commons
Attribution 4.0 International License.
The Dessimoz Lab
blog is licensed under a Creative
Commons
Attribution 4.0 International License.