•
Author: Natasha Glover •
∞
All living organisms descended from a common ancestor, and therefore all living organisms have some genes in common. Therefore, we can take any two species on the tree of life and find out what set of genes were present in the common ancestor of the two species by looking for the genes that they still share to this day. When two species are closely related, i.e. close together on the tree of life, we can observe that much of their genome is the same. But what do I mean by “same”, and what do I mean by “shared”?
Genomes are complex, with many layers of control (DNA-level, RNA-level, protein-level, epigenetic level, alternative splicing-level, protein-protein interaction level, transposable element-level, etc). Because of this complexity, there could be many ways to compare the genomes of two different species to determine which portion of the genome is shared.
In this blog post, when I refer to “shared” parts of the genome, I’m referring to orthologs, which are genes in different species that started diverging due to a speciation event. Orthologs can be inferred from complete genome sequences, and there are many different methods to do this. In particular, I will focus on the Dessimoz lab’s method and database, Orthologous MAtrix, or OMA. OMA uses protein sequences from whole genome annotations to compute orthologs between over 2000 species. The OMA algorithm is graph-based, compares all protein sequences to all others to find the closest between two genomes, allows for duplicates, and uses information from related genomes to improve the calling. OMA is available at https://omabrowser.org/.
It’s quite well-publicized that humans and chimps are extremely similar in terms of gene content, which is why they could be considered our evolutionary cousins1. However, when two species are more distantly related, there is less of their genomes which are the same. But how distant can we go in the tree of life and still see the same genes between different species? For example, how many genes do I (a human) have in common with a plant? Can we even detect orthologs between two species so evolutionarily distant? And if so, what biological/functional role do these genes play?
In this blog post, I will show how to use OMA and some of its tools to find out how much of the human genome we share with plants. There are many different plant species with their genomes’ sequenced, so I will use the extensively-studied model species Arabidopsis thaliana as the representative plant to compare the human protein-coding genes to.
Setup
For the remainder of this blog post, I will show how to use the OMA database, accessed via the python REST API, to get ortholog pairs between two genomes.
In python, I use the requests library to send queries for information to the server housing the OMA database. Pandas is a well-known python library for working with dataframes, so I will use that for analysis. For more information, see:
Let’s start our python code by importing libraries:
import requests
import json
import pandas as pd
The OMA API is accessible from the following link, so we will save it for later use in our code:
api_url = "https://omabrowser.org/api"
Getting pairwise orthologs with the OMA API
Pairwise orthologs are computed between all pairs of species in OMA as part of the normal OMA pipeline. Accessing the data will result in a list of pairs of genes, one from one species and the other from the other species. Any one gene may be involved in several different pairs, depending of if its ortholog has undergone duplication or not.
It is important to note that here we are only using pairs of orthologs. In OMA, we report several types of orthologs, namely pairs of orthologs or groups of orthologs (Hierarchical Orthologous Groups (HOGs). We use the pairs as the basis for building HOGs, which aggregates orthologous pairs together into clusters at different taxonomic levels. Some ortholog pairs are removed, and some new pairs are inferred when creating HOGs. Therefore, the set of orthologs deduced from HOGs will be slightly different than the set of orthologs purely from the pairs.
Here we will use the API to request all the pairwise orthologs between Homo sapiens and Arabidopsis thaliana. OMA uses either the NCBI taxon identifier or the 5-letter UniProt species code to identify species. I will use the UniProt codes, which are “HUMAN” and “ARATH.” To request the ortholog pairs between HUMAN and ARATH, the request would simply look like:
requests.get(api_url + '/pairs/HUMAN/ARATH/')
However, when sending a request that returns long lists, i.e. lists of thousands of genes, the OMA API uses pagination. This means all the results won’t be returned at once, but sequentially page by page, with only 100 results per page. This is a safeguard to make sure the OMA servers don’t get overloaded with requests. Here I show a simple workaround to get all of the pairs.
Get all pairs
genome1 = "HUMAN"
genome2 = "ARATH"
#get first page
response = requests.get(api_url + '/pairs/{}/{}/'.format(genome1, genome2))
#use the header to calculate total number of pages
total_nb_pages = round(int(response.headers['X-Total-Count'])/100)
print("There are {} pages that need to be requested.".format(total_nb_pages))
> There are 128 pages that need to be requested.
Since we want all the pages, not just the first one, we have to use a loop to go through and request all the pages.
#get responses for all pages
responses = []
for page in range(1, total_nb_pages + 1):
tmp_response = requests.get(api_url + '/pairs/{}/{}/'.format(genome1, genome2)+"?page="+str(page))
responses.append(tmp_response.json())
#some basic tidying up so we only have a list of pairs at the end
pairs = []
for response in responses:
for pair in response:
pairs.append(pair)
#Example of a pair
pairs[0]
> {'entry_1': {'entry_nr': 8066469,
> 'entry_url': 'https://omabrowser.org/api/protein/8066469/',
> 'omaid': 'HUMAN00009',
> 'canonicalid': 'NOC2L_HUMAN',
> 'sequence_md5': 'dc91b2521daf594037a6c318f3b04d5a',
> 'oma_group': 840151,
> 'oma_hog_id': 'HOG:0420566.4b.15b.5a.3b',
> 'chromosome': '1',
> 'locus': {'start': 944694, 'end': 959240, 'strand': -1},
> 'is_main_isoform': True},
> 'entry_2': {'entry_nr': 12384097,
> 'entry_url': 'https://omabrowser.org/api/protein/12384097/',
> 'omaid': 'ARATH12334',
> 'canonicalid': 'NOC2L_ARATH',
> 'sequence_md5': 'f19ac310a5e56443f2ce0e4e832addb3',
> 'oma_group': 840151,
> 'oma_hog_id': 'HOG:0420566.1c',
> 'chromosome': '2',
> 'locus': {'start': 7928254, 'end': 7931851, 'strand': 1},
> 'is_main_isoform': True},
> 'rel_type': '1:1',
> 'distance': 138.0,
> 'score': 1094.969970703125}
We can see that the data is in the form of a big list of pairs, with each pair being a dictionary. In this dictionary, one key is entry_1, which is the human gene, and the second key is entry_2, which is the arabidopsis gene. The values for each of these keys are dictionaries with information about the gene. Additionally, there are other key value pairs in the pair dictionary which gives information computed about the pair, such as rel_type, distance, and score (will be explained later).
Make dataframe with all pairs
I personally like to work with pandas because I find it easy to use, so I will import the information about the pairs into a dataframe.
#use pandas to make dataframe
df = pd.DataFrame.from_dict(pairs)
def make_columns_from_entry_dict(df, columns_to_make):
'''Parses out the keys from the entry dictionary and adds them to the dataframe
with an appropriate column header.'''
genome1 = df['entry_1'][0]['omaid'][:5]
genome2 = df['entry_2'][0]['omaid'][:5]
df[genome1+"_"+columns_to_make] = df.apply(lambda x: x['entry_1'][columns_to_make], axis=1)
df[genome2+"_"+columns_to_make] = df.apply(lambda x: x['entry_2'][columns_to_make], axis=1)
return df
#clean up the columns of the entry_1 and entry_2 dictionaries
df = make_columns_from_entry_dict(df, "omaid")
df = df[['HUMAN_omaid','ARATH_omaid','rel_type','distance','score']]
#Here's a snippet of the dataframe
df[:10]
|
HUMAN_omaid |
ARATH_omaid |
rel_type |
distance |
score |
|---|
| 0 |
HUMAN00009 |
ARATH12334 |
1:1 |
138.0 |
1094.969971 |
|---|
| 1 |
HUMAN00029 |
ARATH38493 |
1:1 |
168.0 |
246.330002 |
|---|
| 2 |
HUMAN00032 |
ARATH38034 |
1:1 |
69.0 |
773.869995 |
|---|
| 3 |
HUMAN00034 |
ARATH39850 |
m:n |
140.0 |
635.900024 |
|---|
| 4 |
HUMAN00034 |
ARATH33319 |
m:n |
144.0 |
655.409973 |
|---|
| 5 |
HUMAN00034 |
ARATH01678 |
m:n |
137.0 |
723.580017 |
|---|
| 6 |
HUMAN00034 |
ARATH07547 |
m:n |
134.0 |
761.450012 |
|---|
| 7 |
HUMAN00036 |
ARATH01479 |
1:1 |
103.0 |
554.590027 |
|---|
| 8 |
HUMAN00037 |
ARATH10852 |
1:1 |
60.0 |
2325.889893 |
|---|
| 9 |
HUMAN00052 |
ARATH02626 |
1:1 |
95.0 |
328.959991 |
|---|
Here you can see that each row is a pair of orthologs, with some information about each. The rel_type is the relationship cardinality, between the orthologs. 1:1 means only one ortholog in human found in arabidopsis, whereas 1:m would mean it duplicated in ARATH. m:n means there were lineage-specific duplications on both sides.
How many pairs between HUMAN and ARATH?
print("There are {} pairs of orthologs between {} and {}.".format(len(df), genome1, genome2))
> There are 12792 pairs of orthologs between HUMAN and ARATH.
What percentage is this of the human genome? What percentage of the Arabidopsis genome? To answer this we need to 1) Get the number of genes in each genome that this represents, because there can be more than 1 pair of orthologs per gene. 2) We also need to get the total number of genes per genome.
First we get the number of genes for each species that has at least one ortholog, using the dataframe from above. We don’t have to worry about alternative splic variants (ASVs) because OMA chooses a representative isoform when computing orthologs.
human_proteins_with_ortholog = set(df['HUMAN_omaid'].tolist())
print("There are {} genes in {} with at least 1 ortholog in {}.".\
format(len(human_proteins_with_ortholog), "HUMAN","ARATH"))
arath_proteins_with_ortholog = set(df['ARATH_omaid'].tolist())
print("There are {} genes in {} with at least 1 ortholog in {}.".\
format(len(arath_proteins_with_ortholog), "ARATH","HUMAN"))
> There are 3769 genes in HUMAN with at least 1 ortholog in ARATH.
> There are 5177 genes in ARATH with at least 1 ortholog in HUMAN.
Get the genomes of HUMAN and ARATH
Now we will again use the API to get all the proteins in the genome. (See https://omabrowser.org/api/docs#genome-proteins). Here I will write a function using the same procedure as above to deal with pagination.
#Use the API to get all the human genes
def get_all_proteins(genome):
'''Gets all proteins using OMA REST API'''
responses = []
response = requests.get(api_url + '/genome/{}/proteins/'.format(genome))
total_nb_pages = round(int(response.headers['X-Total-Count'])/100)
for page in range(1, total_nb_pages + 1):
tmp_response = requests.get(api_url + '/genome/{}/proteins/'.format(genome)+"?page="+str(page))
responses.append(tmp_response.json())
proteins = []
for response in responses:
for entry in response:
proteins.append(entry)
return proteins
human_proteins = get_all_proteins("HUMAN")
#Here is an example entry
human_proteins[0]
> {'entry_nr': 8066461,
> 'entry_url': 'https://omabrowser.org/api/protein/8066461/',
> 'omaid': 'HUMAN00001',
> 'canonicalid': 'OR4F5_HUMAN',
> 'sequence_md5': 'df953df7a11ee7be5484e511551ce8a4',
> 'oma_group': 650473,
> 'oma_hog_id': 'HOG:0361626.2a.7a',
> 'chromosome': '1',
> 'locus': {'start': 69091, 'end': 70008, 'strand': 1},
> 'is_main_isoform': True}
It is important to note that this list of genes/proteins includes ASVs for many species. Therefore we need to take care to remove them to have one represenative isoform per gene. We can do this by filtering the list of entries based on the ‘is_main_isoform’ key. Here is a small function to do that.
#get canonical isoform
def get_canonical_proteins(list_of_proteins):
proteins_no_ASVs = []
for protein in list_of_proteins:
if protein['is_main_isoform'] == True:
proteins_no_ASVs.append(protein)
return proteins_no_ASVs
print("HUMAN\n-----")
print("The number of genes before removing alternative splice variants: {}".format(len(human_proteins)))
human_proteins_no_ASVs = get_canonical_proteins(human_proteins)
print("The number of genes after removing alternative splice variants: {}".format(len(human_proteins_no_ASVs)))
> HUMAN
> -----
> The number of genes before removing alternative splice variants: 30700
> The number of genes after removing alternative splice variants: 20152
#Do the same for ARATH
print("ARATH\n-----")
arath_proteins = get_all_proteins("ARATH")
print("The number of genes before removing alternative splice variants: {}".format(len(arath_proteins)))
arath_proteins_no_ASVs = get_canonical_proteins(arath_proteins)
print("The number of genes after removing alternative splice variants: {}".format(len(arath_proteins_no_ASVs)))
> ARATH
> -----
> The number of genes before removing alternative splice variants: 40999
> The number of genes after removing alternative splice variants: 27627
Proportion of the genomes which have orthologs in the other
Now that we have all the necessary information we can see what percentage of the human and arabidopsis genomes are shared, in terms of proportion of genes with at least 1 ortholog in the other species.
print("The percentage of genes in {} with at least 1 ortholog in {}: {}%"\
.format("HUMAN", "ARATH", \
round((len(human_proteins_with_ortholog)/len(human_proteins_no_ASVs)*100), 2)))
print("The percentage of genes in {} with at least 1 ortholog in {}: {}%"\
.format("ARATH", "HUMAN", \
round((len(arath_proteins_with_ortholog)/len(arath_proteins_no_ASVs)*100), 2)))
> The percentage of genes in HUMAN with at least 1 ortholog in ARATH: 18.7%
> The percentage of genes in ARATH with at least 1 ortholog in HUMAN: 18.74%
So the answer to the original questions is that BOTH humans and arabidopsis have 18.7% of their genome shared with each other. A weird coincidence that it turns out to be the same proportion of both genomes!
Conclusion
Using the OMA database for orthology inference, we found that:
- There are 12792 pairs of orthologs between HUMAN and ARATH.
- There are 3769 genes in human that have at least 1 ortholog in arabidopsis.
- There are 5177 genes in arabidopsis that have at least 1 ortholog in human.
- These numbers represent about 19% of both genomes.
Therefore, we can conclude that over the course of evolution since the animal and plant lineages split (~1.5 billion years ago2), about 19% of the protein-coding genes remain and are able to be detected by OMA.
Stay tuned for the next blog post, where I will show what are the functions of these shared genes!
References
- Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005). DOI: https://doi.org/10.1038/nature04072
- Wang, D. Y., Kumar, S. & Hedges, S. B. Divergence time estimates for the early history of animal phyla and the origin of plants, animals and fungi. Proc. Biol. Sci. 266, 163–171 (1999). DOI: 10.1098/rspb.1999.0617
•
Author: Christophe Dessimoz •
∞
The problem in a nutshell
Quantitative Trait Loci (QTL) are regions of a genome for which genetic variants correlate with particular traits. To take a simple example in plants, one might observe that the average seed size (trait) is significantly larger when considering the subset of a population which has a C at a particular position in the genome than a subpopulation with a T.
The reason QTL identifies genomic regions and not precise positions is that neighbouring variants tend to be inherited together. These regions typically contain hundreds of genes, making it difficult to say which one(s) are causal to the trait variation—if any at all (the causal genetic variation(s) can be in non-coding regions too).
Thus, to prioritise candidate causal genes within a QTL region, researchers typically consider previous knowledge on these genes, to see whether a particular gene “makes sense”. In the case of seed size, it might be a gene previously implicated in growth or regulation, or a gene known to influence seed size in a different species. This process is however requires substantial manual interpretation, and is thus labour-intensive and haphazard.
Enter QTLsearch
We realised that our framework of hierarchical orthologous groups, which relates genes across many species, could be extended to integrate QTL results with previous gene function annotations.
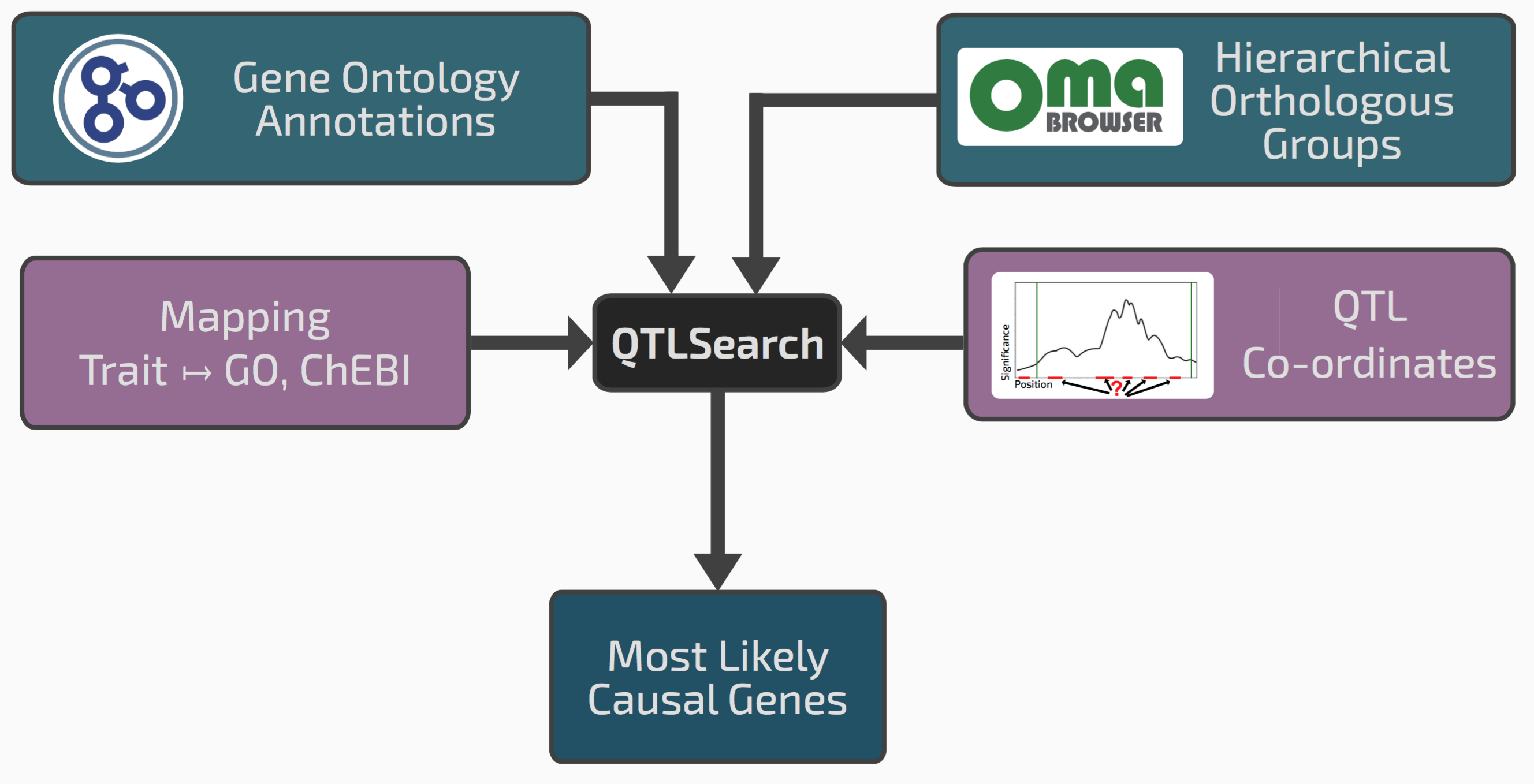
Conceptual overview of QTLsearch
If we go back to the seed size example, it might be that among the genes in the window, one has an ortholog in a different species previously annotated with the GO term “reproductive system development”. This could be a good candidate causal gene.
One risk however in integrating lots of previous knowledge across many species is that we might also find some spurious patterns. We therefore had to devise a way of controlling for random associations between QTL regions and evolutionarily propagated knowledge. Such “null distribution” depends on the specificity or the terms in question, the amount of annotations, the size of the QTL regions, and the species sampling. To cope with this complexity, we chose to implement a non-parametric permutation test.
We implemented the tool as an open source package called QTLsearch, available here.
QTLsearch infers more candidate causal genes than manual analyses
We used QTLsearch to reanalyse two previous studies. In both cases, we could call more candidate genes than the original studies. But more importantly, the evidence behind our calls is fully traceable and statistically supported.
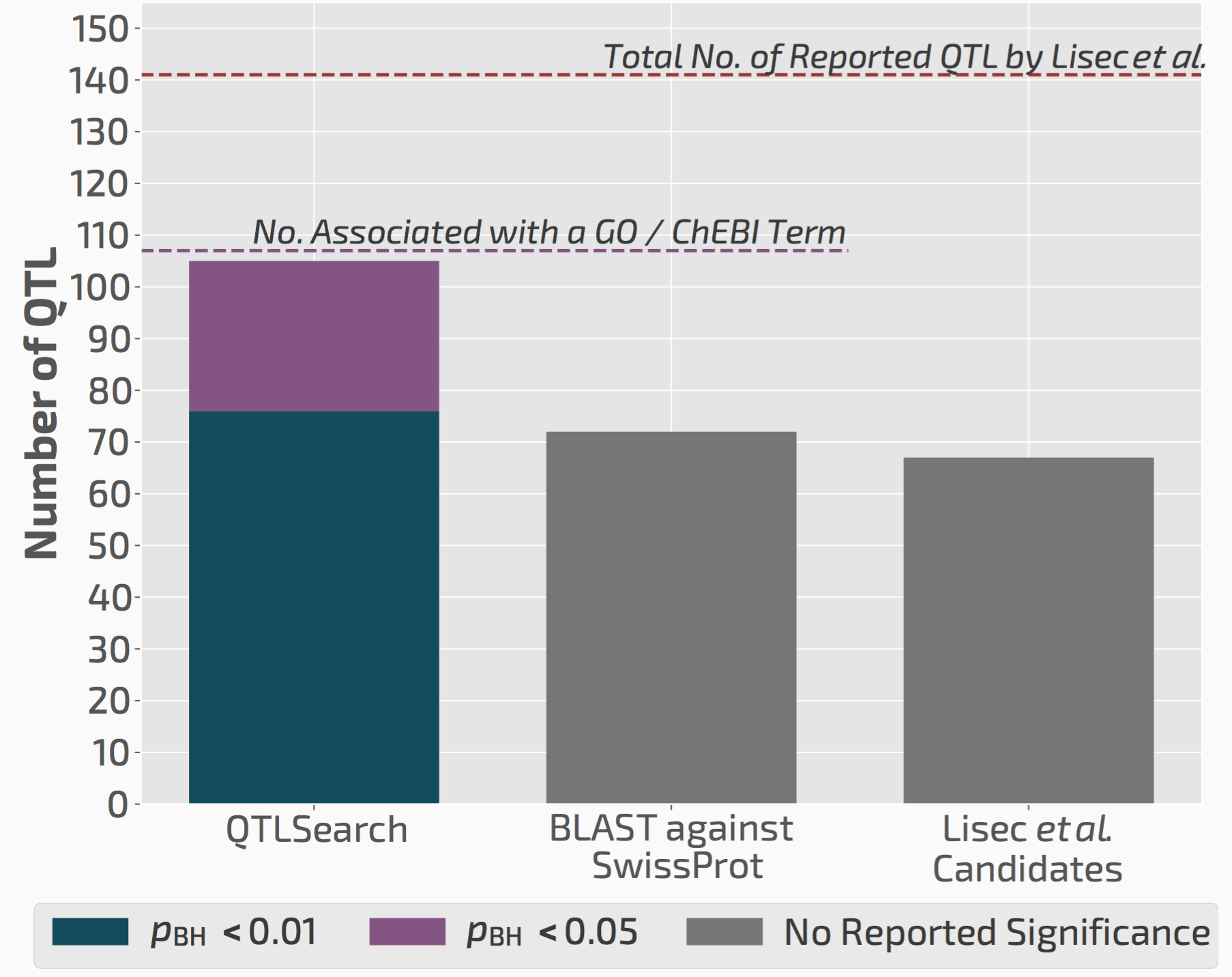
QTLsearch could identify more candidate genes than the original study, but in an automated, reproducible, and statistically meaningful way.
Thus we think this will greatly facilitate future QTL analyses, particularly those that are done in non-model species for which the previous experimental knowledge is very limited.
Behind the paper
This is the third paper that resulted from our collaboration with Bayer CropScience (now BASF CropScience), after our work on homoeologs and on detecting split genes.
The project was conceived by Henning Redestig, collaborator at Bayer at the start of the project (now at DuPont). Henning had contributed to a QTL study and knew how labor intensive the search for putative causal genes is. He realised that HOGs could provide a natural way of integrating functional knowledge across multiple species, to combine the QTL information with previous functional data.
Alex Warwick Vesztrocy, PhD student on the project and first author, ran with the idea—promptly implementing and testing it. Early results looked promising, but Alex soon realised that the mapping between metabolites and GO terms could be improved. He also realised that some terms were quite common, so he devised the approach to compute the significance scores.
Our manuscript was accepted as proceedings paper at the European Conference on Computational Biology (ECCB). In our lab, we like proceedings paper. It’s nice to be able to present the work and publish the paper, particularly since the ECCB proceedings appear in a good journal. More importantly, conferences impose hard deadlines. Deadlines for submission of course, but also for peer-reviewing and for deciding acceptance or not!
Reference
Alex Warwick Vesztrocy, Christophe Dessimoz*, Henning Redestig*,
Prioritising Candidate Genes Causing QTL using Hierarchical Orthologous Groups, Bioinformatics, 2018, 34:17, pp. i612–i619 (ECCB 2018 proceedings)
[Open Access Full Text]